INTRODUCTION
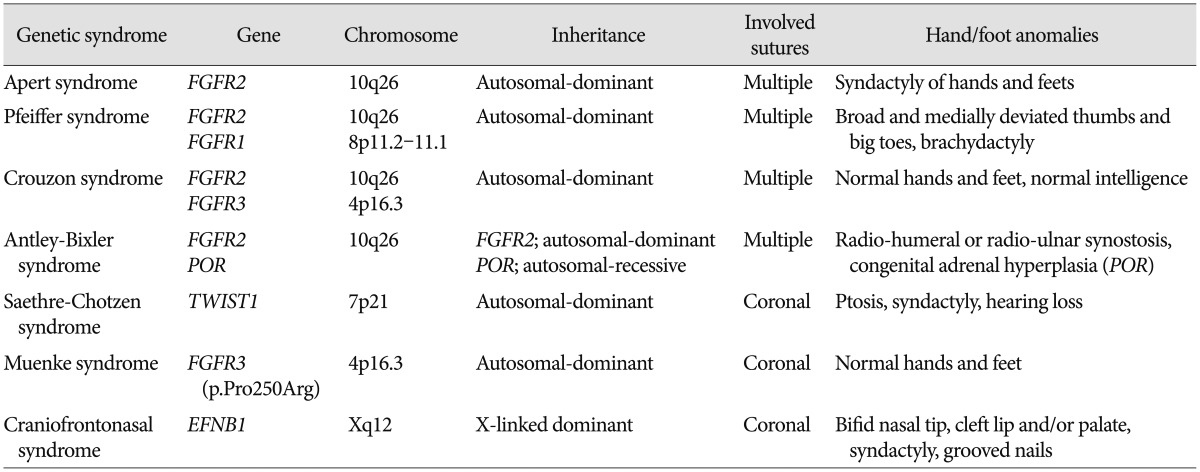
Craniosynostosis describes partial or complete premature fusion of cranial sutures. Ocular hypertelorism, proptosis, beaking of the nose and midface hypoplasia are the common facial features of the craniosynostosis. The prevalence of craniosynostosis is estimated to be 1 in 2100 to 2500 live births11). The sagittal suture (40-55%) is the most commonly affected, followed by the coronal (20-25%), metopic (5-15%), and lambdoid (<5%) sutures13). The syndromic craniosynostosis is the hereditary form of craniosynostosis, which is associated with extracranial phenotypes such as limb, cardiac, central nervous system and tracheal malformations. Syndromic craniosynostosis comprises 15-30% of the total, and specific single gene mutations or chromosome abnormalities could be identified in at least 20% of all cases11,13). Mutations of several genes including FGFR1, FGFR2, FGFR3, TWIST1, and EFNB1 genes have been frequently reported to be associated with syndromic craniosynostosis (Table 1). Among identified genetic causes, mutations of the FGFR2 gene are critically involved in various syndromic craniosynostosis showing multiple suture involvement. However, FGFR2 mutations show variable clinical expressivity, and patients with the same FGFR2 mutation can exhibit diverse clinical manifestations12). Therefore, FGFR2-related craniosynostosis syndromes are usually named according to the accompanying extra-cranial manifestations. Particularly in isolated coronal synostosis, single gene mutations can be detected in one-third of patients13,28). This higher detection rate of mutations may indicate a stronger genetic background for coronal synostosis than other forms of craniosynostosis. Here, common genetic syndromes associated with craniosynostosis are introduced with a brief review of their respective genetic backgrounds.
GENETIC SYNDROMES WITH CRANIOSYNOSTOSIS INVOLVING MULTIPLE SUTURES
Among the various causative genes for craniosynostosis syndromes, FGFR2, FGFR3, and FGFR1 comprise the FGFR family related to craniosynostosis and FGFR2 is the main gene of the family1). FGFRs play a central role in the growth and differentiation of mesenchymal and neuroectodermal cells by binding to FGF and initiation of signal transduction18). Also, FGFRs regulate cranial suture fusion on a macroscopic level. Animal studies suggest that defective FGF signal transduction due to mutations on FGFRs leads to growth arrest of the cranium and the midface20). Mutations in FGFRs have been linked to various clinical craniosynostosis syndromes including Apert (OMIM#101200), Pfeiffer (OMIM#101600), Crouzon (OMIM#123500), Antley-Bixler (OMIM#207410, 201750), Muenke (OMIM#602849), Beare-Stevenson (OMIM#123790), and Jackson-Weiss (OMIM#123150) syndromes. These FGFR-related craniosynostosis syndromes are autosomal-dominantly inherited, and share several craniofacial features including premature closure of multiple cranial sutures. On the other hand, a wide phenotypic range has been shown even in patients with identical FGFR2 mutations17). Therefore, differential diagnosis is usually based on presence or absence of distinct limb and dermatological features18).
Apert syndrome
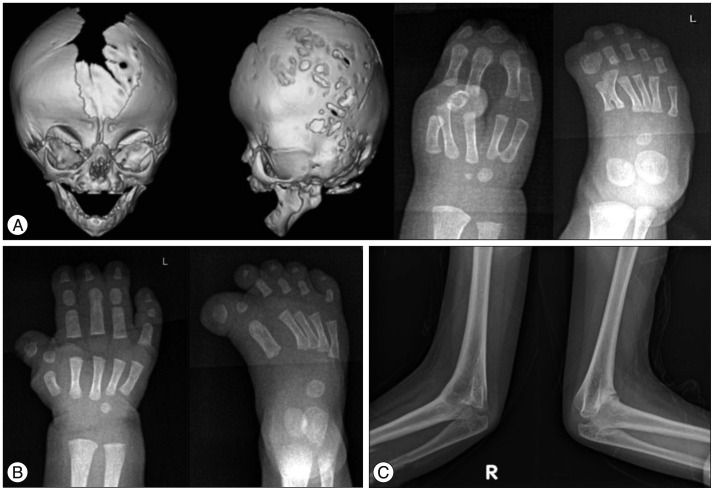
Apert syndrome (AS) is characterized by craniofacial malformations including bicoronal synostosis and severe symmetrical syndactyly of fingers and toes (Fig. 1A). Syndactyly is a characteristic feature of AS that permits distinction from the other FGFR2-related syndromes, and shows a complex fusion leading to 'mitten hand' deformity in both hands and feet. It occurs in 6-15 out of 1000000 live births3). This syndrome is caused by a genetic mutation in the FGFR2 gene, and approximately 98% of all patients have specific missense mutations of FGFR2 located in the linker between the IgII and IgIII domains, either p.Ser252Trp (66%) or p.Pro253Arg (32%)24). Facial manifestations include a flat forehead and retracted midface, proptosis, hypertelorism, and low-set ears. Narrow pharynx and retracted midface frequently result in airway compromise. The other associated anomalies are skeletal malformations, poor joint mobility, eye and ear problems, cleft palate, and orthodontic and other dental problems. Learning disability requiring special education is also commonly accompanied (40-70%)1). Most of patients with AS arise from de novo mutations, which are mainly originated from sperms of their father9).
Pfeiffer syndrome
Distinctive and characteristic features of Pfeiffer syndrome (PS) are broad, radially deviated thumbs and/or big toes along with craniosynostosis. Partial syndactyly on hands and feet can be accompanied in some patients (Fig. 1B). This syndrome affects about 1 in 100000 live births26). Other manifestations including hydrocephalus, proptosis, ankylosed elbows, visceral anomalies, and delayed neuropsychological development may be found. The craniofacial severity is variable among PS patients, and PS can be classified into three clinical subtypes based on the severity of clinical phenotypes. Type 1 has the 'classic' phenotypes with brachycephaly, midface hypoplasia, finger and toe abnormalities, and normal intelligence with generally good outcome. Type 2 and 3 show more severe phenotypes including cloverleaf skull (only shown in Type 3), severe proptosis, elbow ankylosis or synostosis, developmental delay with neurological complications11,26). Mutations in both FGFR2 and FGFR1 cause PS, and FGFR2 mutations found in PS overlap those in Crouzon syndrome. Differentiation between PS and Crouzon syndrome rely on the presence or absence of hands and feet anomalies23,26).
Crouzon syndrome
Crouzon syndrome (CS) is the representative craniofacial dysostosis syndrome, showing a prevalence of 16 in 1000000 live births22). Craniofacial characteristics of CS are a tall and flat forehead, proptosis, and midface hypoplasia. However, the severity of facial deformity is milder than that of AS, and cleft palate is rarely associated with CS. In opposition to AS or PS, CS has normal intelligence, hands and feet14). Most (94%) of CS is caused by mutations in FGFR211), although a specific mutation in FGFR3 (p.Ala391Glu) has been identified in patients with CS and acanthosis nigricans on the skin18). Advanced paternal age linked to de novo development of CS in the offspring has also been demonstrated8).
Antley-Bixler syndrome
Antley-Bixler syndrome (ABS) is a rare form of syndromic craniosynostosis with additional systemic synostosis, including radio-humeral or radio-ulnar synostosis (Fig. 1C). ABS also shows mid-facial hypoplasia, which leads to airway narrowing in most patients. Some patients have congenital heart diseases and renal anomalies. To date, two genes (FGFR2 and POR) have been identified to cause ABS15). Patients with FGFR2 shows severe skeletal manifestations and associated complications without endocrinological or genital abnormalities. However, patients with POR mutations present skeletal manifestations and congenital adrenal hyperplasia with ambiguous genitalia29). In contrast to the inheritance pattern of FGFR2 mutation (autosomal-dominant), POR mutations are inherited in an autosomal recessive fashion. The POR gene encodes P450 oxidoreductase (POR), which transfers electrons to microsomal enzymes, including three other steroidogenic enzymes. Therefore, POR-deficient patients show not only impaired sexual development and steroidogenesis but also skeletal malformations, the mechanism of which is presumed to involve the role of cholesterol biosynthesis in bone formation7).
GENETIC SYNDROMES ASSOCIATED ISOLATED CORONAL SYNOSTOSIS
The coronal synostosis, the second most common form of craniosynostosis, accounts for 20-25% of all patients with craniosynostosis. Single gene mutations can be more frequently detected in one-third of patients with coronal synostosis (bicoronal, 37.5%; unicoronal, 17.5%) than other types of isolated suture synostosis13,28). Among causative genes, the TWIST1, FGFR3, and EFNB1 genes are known to be associated with coronal synostosis, particularly in syndromic patients13).
Saethre-Chotzen syndrome
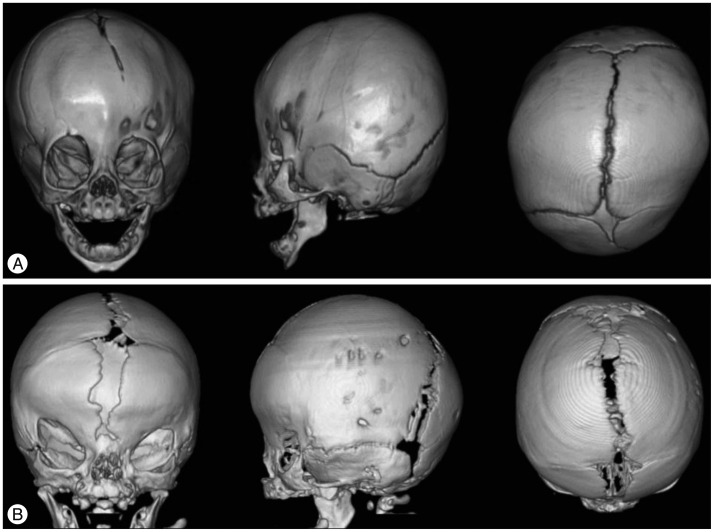
Saethre-Chotzen syndrome (SCS), also known as acrocepha-losyndactyly type III, usually involves unilateral or bilateral coronal synostosis and mild limb deformities (Fig. 2A). It is autosomal-dominantly inherited and is induced by loss-of-function mutations of TWIST1, detected in 60-80% of SCS patients2). TWIST1 encodes for a transcription factor that is responsible for mesenchymal cell development of cranium. TWIST1 also has been thought to be interacted with FGFR2 during fetal developmental6). The estimated prevalence of SCS is 1 in 25000-50000 live births19). The clinical phenotypes of SCS vary profoundly, ranging from isolated unicoronal craniosynostosis to the most extreme manifestation of multiple suture involvement. Other clinical features of this syndrome include ptosis, low-set ears, hearing loss, hypertelorism, broad great toes, clinodactyly, and partial syndactyly. Most patients have normal intelligence21).
Muenke syndrome
Muenke syndrome (MS) is also characterized by unilateral or bilateral coronal synostosis with autosomal dominant inheritance. The other features shown in MS are proptosis, down-slanting palpebral fissures, hearing loss, developmental delay, and specific bone anomalies of the hands and feet24). Significant phenotypic overlap has been detected between SCS and MS, as coronal suture involvement is the major clinical finding. A single mutation of p.Pro250Arg in FGFR3 is the defining molecular characteristic of Muenke syndrome5). However, the mutation, p.Pro250Arg in FGFR3, is also known to be the single largest etiology and it can be found in nonsyndromic cases besides MS4). The birth prevalence harboring this mutation is approximately 1 in 10000 live births, accounting for 8-10% of patients with coronal synostosis16). Considering that MS shows variable expressivity in craniosynostosis and is known as a relatively common diagnosis in patients with craniosynostosis syndromes, testing for p.Pro250Arg in FGFR3 can be recommended as the first-line genetic study to perform in nonsyndromic craniosynostosis patients11).
Craniofrontonasal syndrome
Craniofrontonasal syndrome (CFNS) is a rare form of syndromic craniosynostosis affecting bilateral coronal sutures. Unique facial dysmorphism on the midline structures including hypertelorism, frontal bossing, grooved or bifid nasal tip, cleft lip and/or palate, high arched palate can be the important clues to clinical diagnosis of CFNS (Fig. 2B)1). Clavicle pseudoarthrosis, syndactyly, clinodactyly, broad thumbs with grooved nails, wiry hair, and dental anomalies are also frequently accompanied. Most patients have normal intelligence10). The causative gene EFNB1 on chromosome Xq12, encodes for a membrane-anchored ligand which can bind to an ephrin tyrosine-kinase receptor. This receptor is responsible for the regulation of embryonic tissue-border formation, and is important for skeletal and craniofacial development4). CFNS shows a paradoxical X-linked inherited pattern. Contrary to most X-linked disorders, females are more severely affected in CFNS, whereas males are asymptomatic or show milder facial phenotypes27). This phenomenon is associated with the process of random X-inactivation in females. X-inactivation in females is a process by which one of the two copies of the X chromosome present is inactivated to balance genetic material of the X chromosome with males. The choice of which X chromosome will be inactivated is random. So, mosaic pattern of cells in females may interfere with normal cell-cell interactions and result in the severe clinical phenotypes shown in females25).
CONCLUSION
Great progress in the detection and analysis of craniosynostosis-causative genes has been made in recent years, but craniosynostosis remains a heterogeneous and challenging disorder. Even though specific genetic alterations including single gene mutations or chromosome abnormalities could be identified only in approximately 20% of patients with craniosynostosis, molecular diagnoses can prove helpful in providing adequate genetic counseling to their family members and anticipating associable complications in later life, for these mutation-identified patients.