INTRODUCTION
Neurofibromatosis 1 (NF1) is an autosomal dominant disorder caused by heterozygous mutations of the NF1 gene, which is located on chromosome 17q11.212,14,16). Mutations in NF1 result in loss of neurofibromin, an inactivator of proto-oncogene Ras, leading to increased proliferation and tumorigenesis, therefore, patients with NF1 are predisposed to develop innocent and malignant tumors4,5). In central nervous system, gliomas are the most common neoplasms in individuals with NF119), however, ependymoma with NF1 has rarely been reported. To date, only three cases have been reported in English literature18,21). Moreover, cervical spinal cord ependymoma, to the best of our knowledge, has never been reported occurring in NF1 patients previously. Recently, we experienced a case of cervical spinal cord ependymoma in a patient with NF1. In this report, we discuss the diagnosis, the clinical management, mechanisms of such a rare case with review of the literature.
CASE REPORT
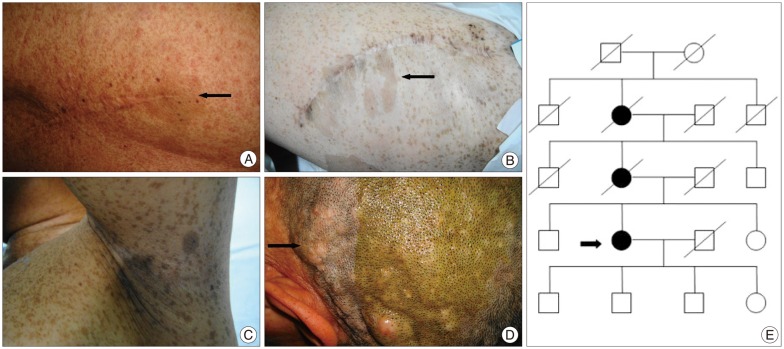
A 49-year-old female patient was admitted to our department because of numbness in her fingers that progressed to her entire body above the bellybutton for half a year. In his past medical history there were expeditiously increscent cutaneous neurofibromas respectively on her back and left thigh for five years, and a total resection for tumors had been performed in a local hospital. Histological examination revealed both of them were neurofibromas with malignant features. On physical examination, widespread caf├®-au-lait spots, axillary and groin frecklings, cutaneous neurofibromas, plexiform neurofibromas and operative scar on her back and left thigh were present, and a sensory deficit was present between the C4 level and bellybutton. No iris hamartomas had been found and mammary gland were normal. Among the family members her mother and maternal grandmother had similar manifestations of NF1, but her daughter and sons had no clinical evidence of NF1 (Fig. 1).
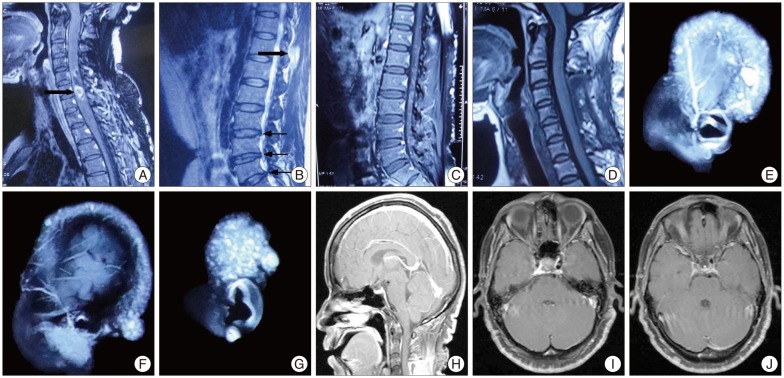
Magnetic resonance imaging showed a relative-demarcated, heterogeneously enhanced mass lesion accompanied by perifocal edema in C5-7 level, a left-sided T11 spinous process heterogeneously enhanced mass in soft tissue, intervertebral disk hernia in L2-5 level and widespread punctum enhancing lesion in her scalp and in T11-L5 level. No evidences of optic pathway glioma and neurofibromatosis 2 (NF2) had been found in head MRI (Fig. 2).
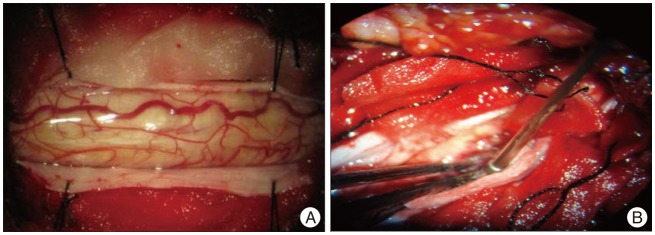
The patient was positioned prone after placement of MEP and SEP monitoring devices, then a posterior laminectomy was performed from C5-7. After confirming the tumor location at the C5-7 level by ultrasonography, we incised the dura at the midline and slit the spinal cord, a yellowish mass with abundance blood supply in the spinal cord was observed. Under an operative microscope, the tumor was gross-totally resected safely, and a laminoplasty from C5-7 was performed at the end of the surgery (Fig. 3).
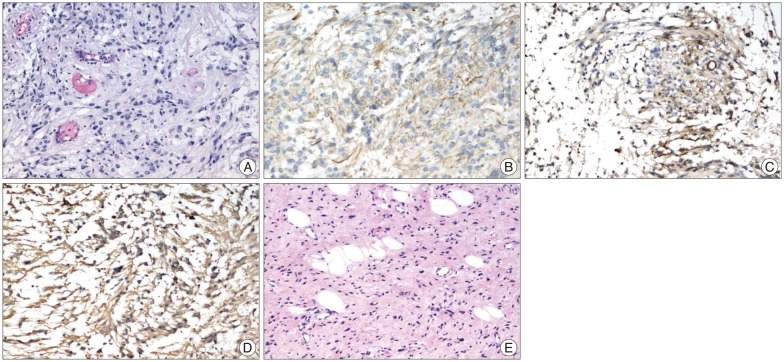
Histological examination revealed it was a moderately cellular glial tumor characterized by round, irregular nucei and eosinophilic cytoplasm, and perivascular pseudorosettes formation was noted. Immunohistochemical analysis showed the tumor cells have immunoreactivity for glial fibrillary acidic protein, epithelial membrane antigen, neuron specific enolase and S-100, but were negative for reticulin. 1% of the cells were positive for monoclonal antibody to Ki-67 antigen. The findings were consistent with ependymoma, grade II in the World Health Organization classification. The biopsy of a scalp neurofibroma showed a typical cutaneous neurofibroma (Fig. 4).
Postoperatively, no adjunctive therapy was given and the patient manifested normal physical development. One year after operation, the neurological disorders had recovered and no neurological deficits were examined. Follow-up magnetic resonance imaging revealed stable postsurgical changes in the C5-C7 level with no evidence of tumor recurrence and dissemination (Fig. 2).
DISCUSSION
Neurofibromatosis 1 occurs with approximately 1 : 2000 to 1 : 5000 in individuals16). The diagnosis of most NF1 patients is based on clinical manifestations. Diagnosis requires at least two major criteria below : 2 or more neurofibromas or 1 plexiform neurofibroma, 6 or more caf├®-au-lait patches, axillary or groin freckling, optic pathway glioma, lisch nodules in the Iris, a distinctive osseous lesion, a first degree relative with NF1 (Table 1)4,8,12). The clinical manifestations of our patient revealed a typical NF1. A considerable history of our patient was malignant cutaneous neurofibromas on her back and left thigh, but the histological examination was not available. In consideration of therapeutic requirement, we performed biopsy of a scalp neurofibroma. The final pathology report revealed neurofibroma without malignant features. Therefore, no specialized treatments for neurofibromas were given.
Spinal cord ependymomas are the most common intramedullary tumors in adults which account for 60% in all spinal cord tumors, and cervical region are the most common localization they occurr3,7,23). The definite diagnosis is confirmed by pathologic findings and the conditions observed in operations. The clinical course of our patient is consistent with spinal cord ependymoma.
Gliomas are often associated with NF1, most with a low grade, mainly locate in the optic nerve10,20), and only 1% in the spinal cord10). However, clinical pathology reports always showed that they were neurofibromas for intraspinal tumors complicated with NF113,24). The incidence of intramedullary gliomas in NF1 patients may be far more than their sporadic counterparts according to similar works17). Meanwhile, compared with NF22,15,25), ependymomas were reported rarely to occur in NF1 patient, to our knowledge, only three cases have been described in English literature19,21). One patient was a 7-year-old boy with NF1 which developed an ependymoma in the left temporal lobe, the tumor was totally removed and histopathological examination revealed a benign ependymoma (grade II in the WHO classification). Fifteen months after the operation, the patient recovered well and MRI showed no recurrence. Another patient was a 12-year-old girl with NF1 which developed a ependymoma in the left temporal lobe, the tumor was also totally removed and histopathological examination revealed a malignant ependymoma (grade III in the WHO classification), although an adjunctive radiotherapy in which 54 Gy was delivered to the tumor site was performed, the tumor recurred and had a widespread transfer, 9 months after the operation, the patient died of an extreme cachexia. The third one was a 55-year-old man with NF1 which developed a thoracic cord ependymoma in T8-L1 level, a laminectomy was performed with good clearance of tumor and histology confirmed a low grade ependymoma. Postoperatively, the patient recovered well and was discharged home with community physiotherapy when a five week course of radiotherapy was completed. In our patient, there were no postoperative neurological deficits, which may benefit from the application of intraoperative ultrasonography and physiological monitoring. As reported, intraoperative ultrasonography and physiological monitoring may reduce a post-operation morbidity in patients1,7,26).
The clinical courses of our patient and others revealed that there were no abnormality between ependymomas in NF1 patients and their sporadic counterparts. Therefore, the management modality is similar, total surgical resection remains the first choice whenever possible3,7,11). In order to reduce the operative morbidity, intraoperative ultrasonography and physiological monitoring should be performed when the ependymoma locates in spinal cord. By the way, the effect of radiotherapy and chemotherapy in ependymoma is uncertain, whether they should be carried out or not is still a controversial subject22). To our patient, no adjunctive therapy has been carried out for the tumor with a low grade and the patient is uneventful. Moreover, NF1 is a multisystem disease, the cooperation between multidisciplinary clinicians and scientists is essential. Whatever the chief complaint presenting from the patient to the doctor, a detailed history talking and physical examination are more fundamental rather than significant managing modality.
Concurrent NF1 glioma mechanism and NF1 genes may closely relate. This gene locates on the long arm of chromosome 17 in the area of 17q11.2, can encode and achieve the synthesis of neurofibromin. This protein as proto-oncogene Ras inhibitors, when NF1 gene function deficiency can result in tumor formation. In addition, NF1 germline mutation, can also lead to tumor form5,12,16). However, specific mechanisms for NF1 complicated with glioma are still unclear, molecular mechanism of tumor gene and potential therapeutic targets for tumor may be becoming the trend and direction of research 4,6,9).
CONCLUSION
Ependymoma with NF1 is a rare situation, we first report a spinal cord ependymoma which occurred in a NF1 patient. Referencing other cases, we find that the clinical course of ependymomas in NF1 patients have no abnormality compared with their sporadic counterparts, therefore, a total-surgical resection remains the first modality whenever possible. The application of intraoperative ultrasonography and physiological monitoring can reduce the operative morbidity. We emphasize that a detailed history talking and physical examination to a NF1 patient are needed and a multidisciplinary cooperation is essential. Further elucidation on the molecular changes of NF1 that drive tumorigeness remains needed aiming to explore a potential therapeutic protocol.