INTRODUCTION
Epilepsy is a chronic neurological disorder affecting many children worldwide. A significant portion of patients have drug-resistant epilepsy that requires a careful work-up and surgical considerations. Focal structural abnormalities in the brain can be defined by high-resolution magnetic resonance imaging (MRI) and other imaging modalities. Extirpation of the lesion provides a high probability of long-term seizure control. Brain tumors are one of the most common structural lesions associated with seizures and epilepsy. As early as 1885, William Osler reported to the Boston Medical and Surgical Journal about a teenage girl who suffered from Jacksonian seizures for 14 years and finally died of status epilepticus [21]. Through an autopsy of her brain, Osler found an almondsized ŌĆśgliomaŌĆÖ under her right motor cortex. Although we do not know the exact diagnosis of the tumor (i.e., the type of glioma) in current nosologic terms, the early age of seizure onset, protracted clinical course, and small nodular features of the lesion suggest that the tumor was a kind of low-grade glioma, such as ganglioglioma. This case confirmed that chronic epilepsy can be caused by a brain tumor near the eloquent cortex and revealed the slow-growing, indolent nature of epilepsy-associated ŌĆśgliomaŌĆÖ.
Our knowledge of brain tumors and epilepsy has greatly evolved since OslerŌĆÖs time. With the advent of neuroimaging techniques, the diagnosis and treatment of brain tumors and epilepsy have been revolutionized. Furthermore, recent advances in molecular genomics urge us to question the basic terms and concepts of neuropathology. For example, what is an astrocyte, and what is astrocytoma? Research has also revealed the hidden nature of epilepsy-associated tumors and their epileptogenic mechanism. This article aims to provide an overview of the evolving landscape of epilepsy-associated tumors, focusing on the pathological and molecular characteristics. The development of and challenges related to the treatment of epilepsy-associated tumors are also considered.
LONG-TERM EPILEPSY-ASSOCIATED BRAIN TUMORS (LEATS)
Brain tumors are the second most common structural lesion associated with epilepsy. Data from the European Epilepsy Brain Bank (EEBB), which contains 9532 epilepsy surgery specimens showed that brain tumors constituted 23.6% of the causative diagnoses, next to only hippocampal sclerosis (36.4%) [6]. Virtually all brain tumors can elicit seizure attacks that, if recurrent, lead to epilepsy. Actually, seizure is one of the major symptoms of meningiomas [3], and even cerebellar tumors can be the source of seizures [23]. However, intra-axial brain tumors involving the cerebral cortex are more closely related to epilepsy than extra-axial tumors or deep-seated intra-axial tumors. Many studies have consistently shown that low-grade brain tumors are more likely to cause epileptic seizures than high-grade brain tumors such as glioblastoma [37]. High-grade tumors tend to damage surrounding structures (including potential epileptogenic areas), whereas low-grade tumors may displace or distort nearby neurons and neuronal networks, evoking seizures.
Some low-grade brain tumors are more frequently involved in chronic intractable epilepsy. Hence, the concept of so-called LEATs was introduced by the Bonn Epilepsy Center [31]. Ganglioglioma and dysembryoplastic neuroepithelial tumor (DNET) are representative tumors of LEATs. Ganglioglioma alone accounted for 10.4%, and DNET constituted 5.9% of the EEBB cohort [6]. Minor subtypes of LEATs include pleomorphic xanthoastrocytoma (PXA), angiocentric glioma, and isomorphic astrocytoma. LEATs, typically ganglioglioma and DNET, are characterized by early age of diagnosis (children and adolescents), the predilection to temporal lobe, cortical involvement, and low probability of malignant progression. Most LEATs are so-called glioneuronal tumors that exhibit both glial and neuronal differentiation patterns. The World Health Organization (WHO) classification of brain tumors also places individual LEATs in mixed neuronal and glial tumor group (but the WHO never officially accepted the term LEATs) [1]. One notable characteristic of LEATs is their close relationship with brain cortical dysgenesis. An original description of DNET was a composite tumor consisting of a tumor with specific glioneuronal elements and an adjacent focal cortical dysplasia (FCD) (so-called ŌĆścomplex typeŌĆÖ) [16]. Later, a simple type with only glioneuronal elements and nonspecific variants (although highly controversial) without glioneuronal elements was added to the DNET category [15,17]. The coexistence of FCD is also commonly observed in ganglioglioma [42]. It is also intriguing that some patients develop a composite neoplasm consisting of DNET and ganglioglioma together [43]. Therefore, the current classification of brain cortical malformation regards DNET and ganglioglioma as a kind of brain malformation (malformations secondary to abnormal neuronal and glial proliferation), alongside FCD and hemimegalencephaly [2]. The frequent temporal lobe involvement, cortical location, low-grade pathology, and association with FCD all appear to contribute to the high propensity of LEATs to cause chronic drug-resistant epilepsy.
Although relevant in clinical practice, several aspects of the concept of LEATs have been questioned. First, the term was originally applied to brain tumors associated with long-term (usually >2 years), drug-resistant epilepsy [31]. However, since the time that the term was proposed, the definition of medical intractability in epilepsy has become less stringent. In particular, in pediatric epilepsy, the strategy of an early imaging work-up and surgery, if possible, has been encouraged to prevent the neurocognitive deficits caused by recurrent seizures and the resulting prevention of normal development [24]. In this regard, changing the phrase ŌĆślong-termŌĆÖ in LEATs to ŌĆślow-gradeŌĆÖ has been proposed, as the majority of LEATs are truly low-grade neoplasms [4].
Second, LEATs are not an established concept. PXAs, once included in LEATs, are now considered to be diffuse gliomas (as opposed to LEATs), mainly because PXA has far more aggressive biological behavior than typical LEATs. From a prognostic perspective, one fourth of PXAs are of a high grade (WHO grade III or IV) and the 10-year overall survival of PXA patients was only 67% [39]. Angiocentric glioma is a newly adopted member of the WHO classification of brain tumors. Angiocentric glioma develops in the brain cortex of children, resembles ependymoma histologically, and has proven to be highly epileptogenic, designating the tumor as a legitimate LEAT [35]. However, angiocentric glioma is a rare tumor with an unknown overall incidence. So-called isomorphic astrocytoma is an intriguing entity with distinct histopathology and excellent prognosis compared with ordinary diffuse astrocytoma, but its validity as a separate entity has not been accepted by neuro-oncology societies [5].
Furthermore, significant pathological and clinical heterogeneity exists in each tumor category. DNETs are highly likely to be associated with chronic epilepsy, but many patients with DNET exhibit only sporadic seizures, no seizures, or even no symptoms. The majority of gangliogliomas are benign, lowgrade tumors, but some tumors exhibit anaplasia in histology and become frankly malignant over the clinical course. Therefore, although widely accepted by epilepsy societies, LEATs have never been formally included in the WHO classification of brain tumor pathology. This discrepancy between clinical features (chronic epilepsy) and pathological coherency (diverse entities) may be compromised by novel findings in genomic studies on LEATs, which will be discussed in a later section.
DIFFUSE LOW-GRADE GLIOMA (DLGG) AND EPILEPSY
DLGG includes astrocytoma and oligodendroglioma. These tumors commonly develop in young adults. DLGG involves large areas of brain cortex and subcortical areas, most notably the frontal lobes. Seizure and epilepsy are the most common manifestations of DLGG. Nearly 80-90% of patients with DLGG develop seizures [36,37]. The so-called ŌĆślow-gradeŌĆÖ glioma incorporates both DLGG and LEATs. The majority of DLGGs correspond to histopathological grade II tumors with a far higher rate of infiltration, recurrence, and malignant transformation than typical LEATs, which are mostly grade I tumors.
Frequently, DLGG has been excluded from the discussion of epilepsy-associated tumors (the EEBB cohort mentioned above does not include DLGG cases) because DLGG is considered a bona fide invasive glioma that should be dealt with in the oncology field [6]. However, many patients with DLGG attain long-term survival and subsequently face the same problem of long-standing epilepsy as patients with LEATs. In fact, any epilepsy surgery cohort must include a number of patients with DLGG in addition to the backbone of the LEAT group, especially when adolescents and young adults are included [40]. To make the situation more complex, DLGG can be roughly divided into so-called adult-type tumors and pediatric-type tumors, according to the mutation profiles. For example, pediatric-type astrocytomas lack IDH1/2 mutations and rarely undergo malignant transformation. So-called pediatric-type oligodendrogliomas are frequently devoid of IDH1/2 mutation and 1p/19q codeletion, the hallmarks of oligodendroglioma [29,45]. A study re-evaluated 100 cases of histologically-diagnosed oligodendrogliomas from patients under the age of 20 years [48]. In the study, a pathology review revealed that only 50 cases were considered oligodendrogliomas and the others were reclassified as DNET, gangliocytoma, ependymoma, and astrocytoma. In the 50 cases of oligodendrogliomas, IDH1 mutation and 1p/19q codeletion were found in only 18% and 25% of cases, respectively [48]. It is difficult to diagnose tumors with oligodendrocyte-like cells because many low-grade gliomas share histologic features. There is certainly a fair amount of overlap between the pediatric-type DLGGs and LEATs on pathological and clinical grounds. Therefore, to evaluate the true pathological spectrum of epilepsy-associated tumors, molecular classification should be considered.
Clinically, oligodendrogliomas in children tends to be smaller and more commonly found in the temporal lobe than their adult counterparts which develop most frequently in the frontal lobe [22]. Pediatric oligodendrogliomas has far better prognosis than adult tumors of the same histology [22]. The different molecular characteristics of pediatric-type and adult-type DLGG may underlie this phenomenon. Erroneous classification and inclusion of LEATs (esp., DNET and ganglioglioma), as discussed above, may also contribute the better survival of pediatric patients.
MOLECULAR LANDSCAPE OF EPILEPSY-ASSOCIATED TUMORS
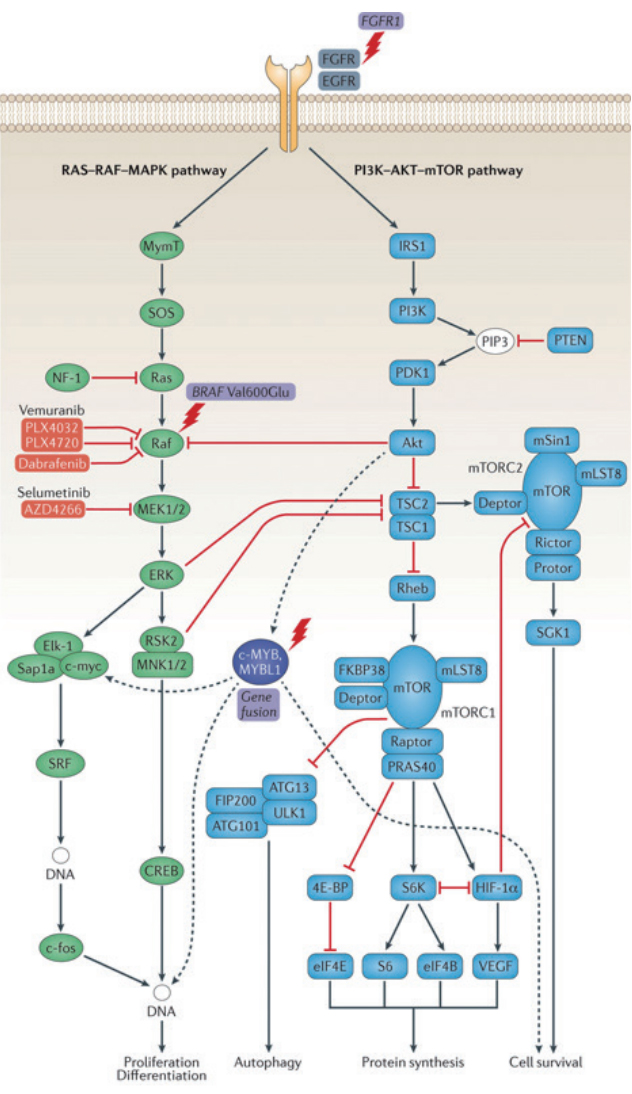
Recent rapid progress in sequencing technology and the refined classification based on genomic signatures have allowed unprecedented opportunities to understand brain tumors, especially low-grade gliomas. In pediatric low-grade gliomas, whole genome sequencing revealed that most pediatric low-grade gliomas have alterations of BRAF, FGFR, and MYB/MYBL1 genes [54]. In particular, pediatric DLGG showed either duplication of the tyrosine kinase domain of the FGFR1 gene or rearrangement of the MYB/MYBL1 genes. Mutations of IDH1/2, the hallmark of adult-type DLGG were rarely found in pediatric DLGG, supporting the distinction of pediatrictype DLGG from its adult counterpart [46,54]. Mutations of TP53 and ATRX, the other common variations in adult-type diffuse astrocytomas were also rarely found in pediatric-type DLGG.
In LEAT group, alterations of the BRAF gene, notably the BRAF V600E mutation is highly represented in gangliogliomas [18]. In 36 gangliogliomas that were sequenced, 27 tumors (75%) had a BRAF V600E mutation, variant BRAF mutation, or BRAF fusion. The other tumors had mutations in KRAS, RAF, NF1, or an FGFR1/2 alteration, confirming that ganglioglioma develops through alterations in the RAS-RAF-MAPK pathway [38]. A study showed that mutated BRAF V600E protein was predominantly expressed in neuronal lineage cells, although BRAF was expressed in both the neuronal and glial compartments [27]. Interestingly, when the BRAF V600E pathogenic mutation was introduced into mouse embryos, the embryos postnatally developed an increased neuronal size and epileptic phenotype, whereas the same mutation transfected into postnatal mouse brains led to glial proliferation only, indicating that BRAF V600E induced epileptogenesis in the neuronal lineage and tumorigenesis in the glial lineage [28].
A large-scale study on DNET revealed that alterations of FGFR1 (duplication or point mutation) were predominant (58%) with no observed cases of BRAF alterations [47]. Previous small-scale studies reported the presence of BRAF alterations in up to 30% of DNET [8,34]. The discrepancy in BRAF alterations can be explained by the fact that examining ŌĆśnon-DNETŌĆÖ (DNET tissues that do not meet the strict WHO criteria in a central pathology review) revealed a lower frequency (19%) of FGFR1 mutations and a higher proportion (13%) of BRAF V600E mutations [47]. The interobserver reliability of histopathological diagnoses of LEATs has been questioned [50]. Many glioneuronal tumors lack specific histological features that are crucial for the diagnosis of ganglioglioma or DNET, making diagnosis difficult. Immunohistochemistry with staining for CD34, S100, and synaptophysin is helpful, but the markers are essentially nonspecific [51]. Some researchers have tried to extend the definition of DNET to include these nonspecific, aberrant glioneuronal tumors [17]. Recently, Stone et al. [50] found that LEATs were roughly divided into two molecular groups, regardless of their histology. After a histologic review of archived LEAT tissues, they reclassified them into three groups : ganglioglioma, DNET, and glioneuronal tumor not otherwise specified. Expression and methylation profiling segregated all cases into two molecular groups, one predominantly with ganglioglioma (group 1) and the other mainly with DNET (group 2). It is noteworthy that each group included both ganglioglioma and DNET in different proportions. As expected, group 1 was enriched for the BRAF V600E mutation, and group 2 was enriched for FGFR1 alterations. Qaddoumi et al. [44] also reported similar findings: diverse low-grade glioneuronal tumors were segregated into BRAF-group/ganglioglioma-like tumors, FGFR1 group/tumors with oligodendrocyte-like cells (including DNET), and MYB group/astrocytoma-like tumors. A small but non-negligible overlap existed between the molecular groups. These studies revealed that the histological differentiation of LEATs (and DLGG) is incomplete and that molecular profiling can reveal the fundamental characteristics of the tumors, enabling more precise diagnosis and classification.
TREATMENT OF TUMOR-RELATED EPILEPSY IN THE MOLECULAR ERA
In tumor-related epilepsy, treatment should have two aims : tumor control and seizure freedom [40]. Because the majority of LEATs are grade I tumors, complete surgical resection confers excellent long-term tumor control. According to the Surveillance, Epidemiology, and End Results (SEER) database of pediatric patients with ganglioglioma/gangliocytoma, who were treated between 2004 and 2010, two-thirds of patients received gross total resection (GTR). The five-year overall survival of patients with non-brainstem tumors was 96.6% [19]. Despite excellent overall survival, progression-free survival was far lower (37% at 10 years in one study), indicating that recurrence is not uncommon [13]. It should be noted that ganglioglioma has some malignant potential, usually after recurrence, although it is rare in children [26,33]. GTR is the most important factor for long-term tumor control of ganglioglioma, as in other low-grade tumors. Previously, it was suggested that DNET has a low likelihood of recurrence even after subtotal resection [16]. However, recent studies have indicated that DNET has a higher recurrence potential than previously thought [14,32]. Some DNETs have poor delineation and gray-white matter blurring around the tumor on MRI, indicating the existence of adjacent FCD [10]. GTR of this type of DNET has proven difficult [9]. In a meta-analysis of DNET treatment, the median GTR rate was 79% (interquartile range 62-92%) [7]. Many studies did not report the actual recurrence rate as documented on MRI but only mentioned the reoperation rate, which ranged from 5-46% [7]. DNET tends to grow very slowly even after recurrence, frequently without symptoms. Reoperation is also inadequate to evaluate the true oncological properties of DNETs, because reoperation may be necessary to control seizures regardless of tumor recurrence. In a study evaluating the radiological recurrence of LEATs, the long-term recurrence-free survival of DNET was approximately 75% [52].
Seizure freedom after surgery was variable, but the data suggested that there was 80-85% long-term (>5 years) seizure control [20,31,40]. GTR of the lesion was also an important factor affecting seizure control, but other clinical factors, such as shorter duration of epilepsy or the absence of secondary generalization, were associated with better outcomes in some studies [20]. Although factors affecting seizure control are more complex than tumor control, it seems obvious that the two outcomes are very closely related. Seizure recurrence is commonly the first sign of tumor recurrence. Pallud and McKhann [37] provided relevant examples in which seizure frequency was attenuated according to a decrease in tumor burden. Seizure recurrence was significantly more frequent for DNET than for ganglioglioma [52]. Seizure control seems less favorable for DLGG than for LEATs. Long-term seizure control for diffuse astrocytoma was 66% in the Bonn series [31]. In the Beijing series, one-year seizure freedom was 65% after resection of DLGG [53]. The former study included both pediatric and adult patients, and the latter series is composed of only adult patients. The median duration of epilepsy was far longer in the Bonn series than in the Beijing series (12 years vs. 10 months), reflecting the different tumor biology of the subjects of each study. The unfavorable seizure outcome of DLGG may be attributed to the diffuse, invasive nature of DLGG and the higher probability of incomplete resection [31].
There has long been an unsettled controversy over whether tumor resection alone (lesionectomy) or extended lesionectomy (epilepsy surgery including nearby epileptogenic cortex) yields superior seizure control. The surgical strategy differs greatly from center to center, and there is no clear evidence supporting either strategy, partly because the patients and pathologic spectrum of tumor-related epilepsy are too heterogeneous [40]. The Bonn series showed that their surgical procedure changed from a standard temporal lobectomy to more tailored type lesionectomy over the years, and the seizure outcome improved during the same period [31]. Other studies also reported no or very few advantages of extended lesionectomy over tumor resection [12,20,52]. Intraoperative electrocorticography (ECoG) may be useful for detecting the epileptogenic cortex, but the impact on seizure outcome is variable [20,49]. It appears that surgical outcome depends more on the pathology than on the choice of surgical procedure if the GTR of the tumor is guaranteed. Some LEATs are not easily differentiated from FCD on MRI. Because epilepsy surgery using invasive monitoring and ECoG is usually required for FCD, differential diagnosis is important. 11C-methionine positron emission tomography (PET) can be helpful because ganglioglioma and DNET show higher methionine uptake than FCD [11,41].
Many pediatric brain tumors have only one driver mutation. As we have seen, epilepsy-associated tumors, including many LEATs and pediatric-type DLGG, have a key driver mutation in BRAF, FGFR1, or MYB/MYBL1. BRAF is a canonical member of the RAS-RAF-MAPK pathway. FGFR1 is an upstream receptor tyrosine kinase of this pathway, and MYB and MYBL1 are also thought to be involved in the regulation of this signaling pathway (Fig. 1) [4]. There is evidence that the BRAF V600E mutation acts on neuronal cells of ganglioglioma to promote epileptogenesis [28]. It is also interesting that the highly epileptogenic FCD is largely driven by somatic mutations of the PI3K-AKT-mTOR pathway, with which FGFR1 and MYB/MYBL1 are also involved [4,30]. Many drugs targeting the RAS-RAF-MAPK pathway, especially BRAF inhibitors, are in clinical trials for the treatment of low-grade gliomas [25]. Although the oncological effect of BRAF inhibitors is questionable as of yet, their efficacy for seizure control in residual BRAF-mutated glioma has not been addressed and needs future evaluation.
CONCLUSION
Epilepsy-associated tumors are generally studied in the context of low-grade gliomas due to the high frequency of seizures as symptoms of the diseases. The f lorid emergence of new entities of glioneuronal tumors and the concept of LEATs has greatly expanded our understanding of the clinical and pathological spectrum of tumor-related epilepsy. Recent development in genome research have further widened the horizon of epilepsy-associated tumors. Low-grade gliomas can be divided into LEATs, pediatric type DLGG, and adult-type DLGG. LEATs and pediatric type DLGG are characterized by major mutations in BRAF, FGFR1, and MYB/MYBL1. Furthermore, mutation-centered classification (also expressionand methylation-based approaches) may be more accurate in the diagnosis of these tumors because the histopathological differentiation can be difficult in many LEATs and in DLGG. Targeted therapy against the oncogenic driver mutations may be applied not only for tumor control but also for seizure control.