INTRODUCTION
Stroke, one of the most common clinical cardiovascular and cerebrovascular diseases in China, is characterized by high morbidity (50%) and disability rate (20%), even become life-threatening in severe cases [35]. Stroke is a complex neurological disease caused by multiple factors, including unchangeable factors (genetic factors) and changeable factors (environmental factors). However, none of these mechanisms can fully explain the pathogenesis of stroke. Epidemiological studies have extensively considered that the occurrence of stroke has a greater relationship with genetic factors [19]. Based on the National Stroke Epidemiological Survey (China), the incidence rate of stroke and the age-standardized incidence rate among Chinese residents had been reported to be 345.1/100000 and 246.8/100000, respectively, in 2013 [31]. The incidence of ischemic stroke (IS) and hemorrhagic stroke in China, as shown by the 2016 Global Burden of Diseases, was 276.75/100000 and 126.34/100000, respectively [14]. In recent years, amounts of researches have revealed that the incidence of IS in China has consistently increased.
In China, the incidence of stroke has exhibited sustained growth within the last three decades. With the acceleration of aging population and urbanization as well as unhealthy lifestyle, risk factors for stroke are generally exposed, and the incidence of stroke has risen sharply. The burden of stroke in our country exhibits an explosive growth trend and a significant trend of rapid growth among low-income groups, obvious gender and regional differences as well as getting younger. It is estimated that the incidence of cerebrovascular disease events in 2030 will be about 50% higher than that in 2010 [16,23]. IS accounts for more than 85% of stroke, and has a trend of younger age. Moreover, IS with high morbidity, disability, recurrence, and death rate. IS has been confirmed to cause a series of complications such as permanent neurological deficit, disability and even death in patients. In addition, it may also lead to dementia in the elderly, senile epilepsy and post-stroke depression [29], which has exerted a great burden on country, families and society, making it an important public health problem. Therefore, it is of great significance to further explore the mechanism of the occurrence and development of IS, and to find effective predictors and therapeutic targets.
Bioinformatics analysis is an important part of molecular biology and it is widely used to associate diseases with genes, explain the pathogenesis of diseases, and lay the foundation for the diagnosis, treatment and prognosis of diseases [27]. The GEO database of the National Center for Biotechnology Information (NCBI) is considered the most comprehensive public repository of large-scale genomics data, which can not only efficiently store high-throughput functional genome data, but also provide complete and well-annotated data storage. The original data are extracted from GEO database, and the differentially expressed genes (DEGs) are screened by bioinformatics software and methods. The molecular mechanism of DEGs regulating IS metastasis was further analyzed by using Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), and database annotation, visualization and integrated discovery (DAVID). The protein-protein interaction (PPI) network is constructed by using the search tool of searching interactive gene (STRING) database and Cytoscape software, and the Hub gene is identified according to the core modules.
The purpose of the present study was to analyze the molecular mechanism of IS by bioinformatics methods, to find the key genes of stroke, and to provide reference information for the diagnosis, treatment and evaluation of IS.
MATERIALS AND METHODS
The research was under the approval of the Ethics Committee of The First Affiliated hospital of Dali University.
Search and download of microarray data
The microarray data was downloaded from the GEO database (http://www.ncbi.nlm.nih.gov/geo/) with the keywords ŌĆ£ischemic strokeŌĆØ. The selection criteria of CHIP contained : 1) sequencing blood samples derived from clinical IS patients, excluding animal sources; 2) enrolled studies all had control groups; and 3) the chip platform included gene type and gene ID information. Two data sets, GSE58294 and GSE22255, were finally selected based on the above criteria. The data platform of GSE58294 was based on GPL570, including 69 cases in IS group and 23 cases in normal control group. The data platform of GSE22255 was based on GPL570, including 20 cases in IS group and 20 cases in normal control group.
Identification of DEGs
The IS group and control group samples of the two data sets was analyzed using GEO2R online analysis tool (http://www.ncbi.nlm.nih.gov/geo/geo2r), and the DEGs were obtained respectively. The screening criteria for the DEGs were as follows : p-value <0.05, |log2FC| >0.5). The DEGs with a logFC >0 were considered up-regulated genes, while the DEGs with a logFC <0 were considered downregulated genes. The intersection of genes with up-regulated and down-regulated expression in the two data sets were taken, and the co expressed up-regulated or down-regulated genes were screened. Funrich Software (version 3.0; http://funrich.org/index.html) was utilized to generate the Venn diagram of DEGs.
Functional enrichment analysis
GO and KEGG pathway enrichment analysis were used for DEGs using the DAVID database (https://david.ncifcrf.gov/) [13], and the biological process (BP) analysis and KEGG signal pathway results of DEGs were obtained. The GO covered three domains : BP, cellular component (CC), and molecular function (MF). p<0.05 was determined as statistically significant.
PPI network construction
The DEGs were input into STRING database (https://string-db.org/) to construct PPI network.
PPI networks were constructed using the STRING obtained Cytoscape (https://cytoscape.org). PPI information obtained in the STRING was analyzed by Cytoscape, and the core targets in the PPI network were determine by Cytohubba software according to the centrality score for screening for core pathogenic genes.
Hub gene related-microRNA (miRNA)
The top 5 hub gene were mapped to corresponding miRNAs using NetworkAnalyst 3.0 (https://www.networkanalyst.ca/). The NetworkAnalyst 3.0 is a visual online platform used for finding miRNA-gene interactions in Gene Regulatory Networks. The hub genes-miRNA network maps were constructed using Cytoscape ver. 3.7.2.
RESULTS
Identification of DEGs in IS
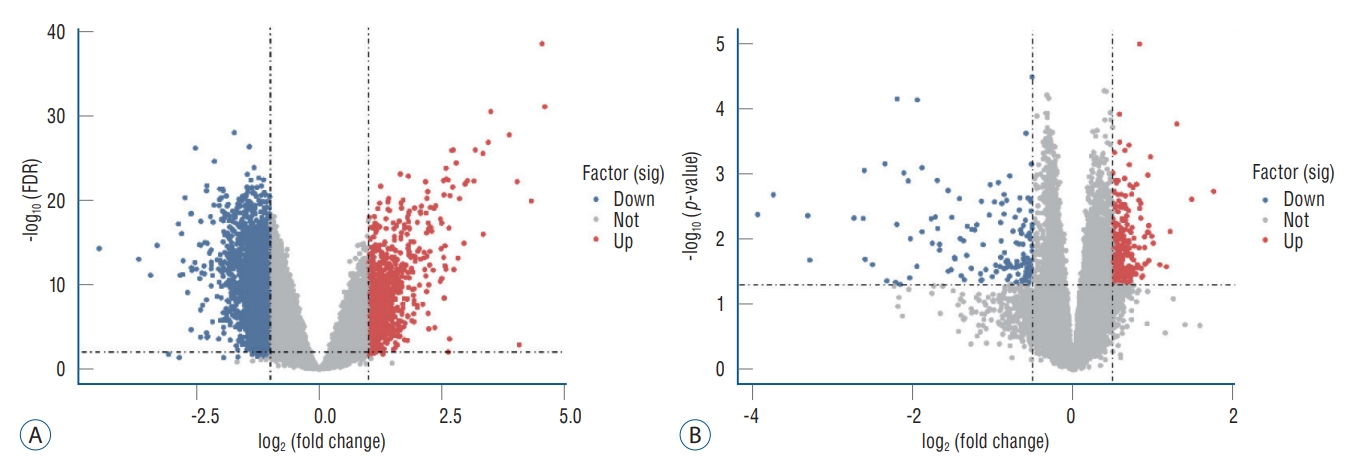
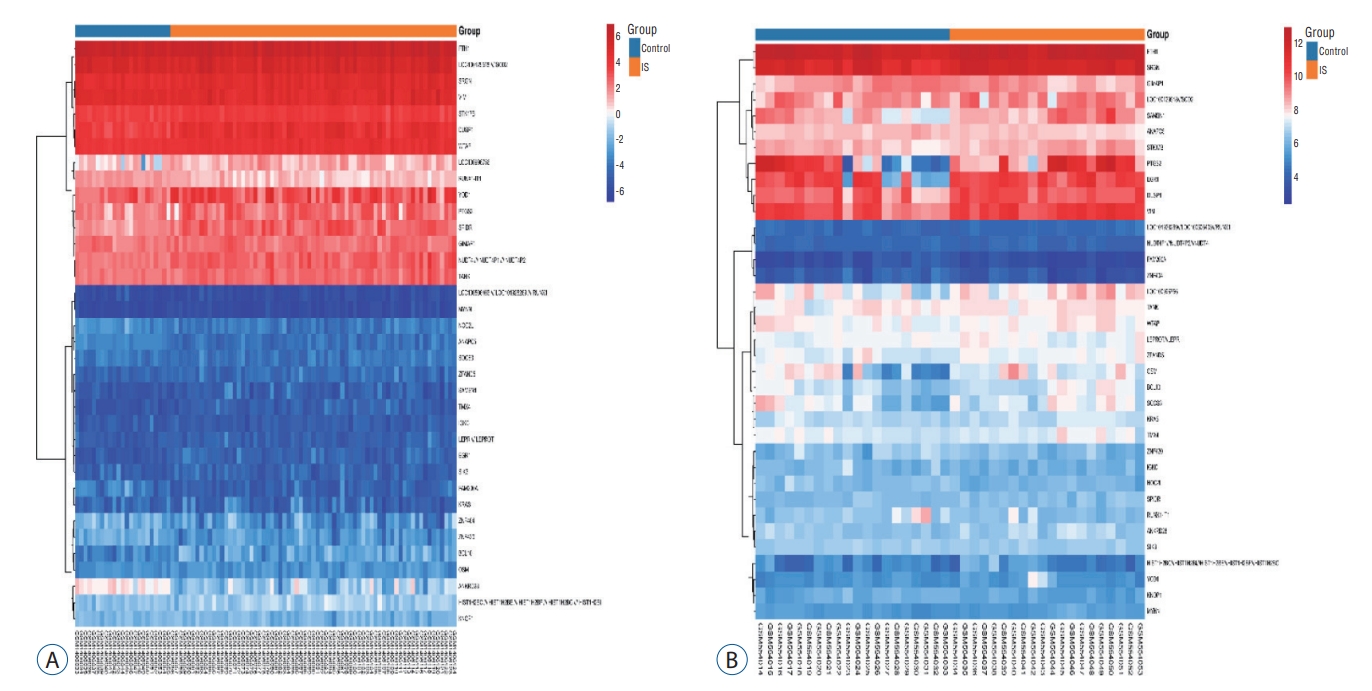
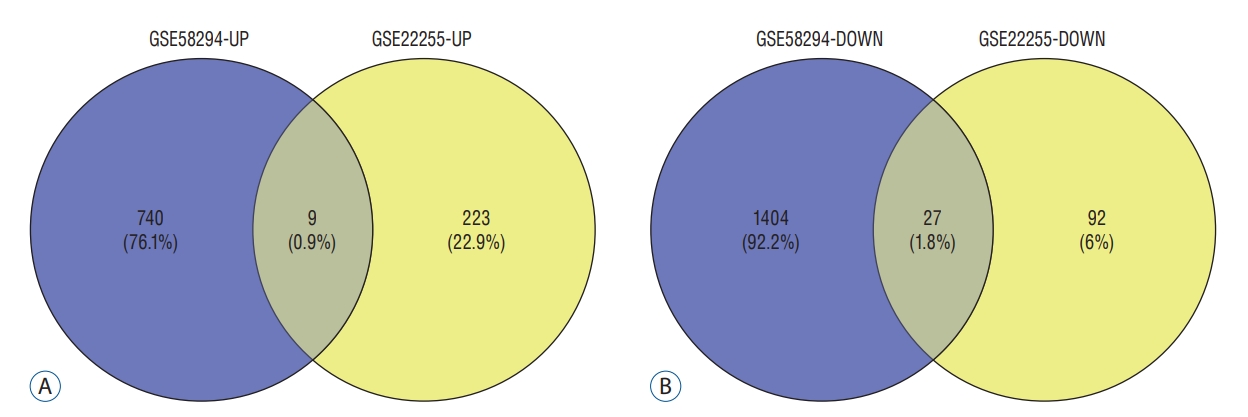
According to the screening criteria, IS related expression data sets GSE58294 and GSE22255 were extracted from GEO database. According to the criteria of p<0.05 and |log2FC| Ōēź0.5, there was a total of 981 DEGs in GSE58294, including 749 up-regulated genes and 232 down-regulated genes in DEGs respectively. A total of 1550 DEGs in GSE22255, including 1341 up-regulated genes and 119 down-regulated genes in DEGs respectively. The gene expression profiles of two DEGs containing two sets of sample data were shown in Fig. 1. Cluster analysis was performed on the chip information to construct the heatmap (Fig. 2), and the results showed that there were differences in the expression of DEGs in IS. The aforementioned genes were further screened and analyzed by Venn diagram. As shown in Fig. 3, 36 genes were significantly differentially expressed between the two groups, of which 27 genes were up-regulated and nine genes were down-regulated, demonstrate in Table 1. The expression of GSE58294 ranged from -6 to 6, and the expression of GSE22255 ranged from 2 to 12. The variation range of the two data sets was relatively small, and the results were shown in Fig. 4.
GO functional enrichment analysis and KEGG pathway enrichment analysis
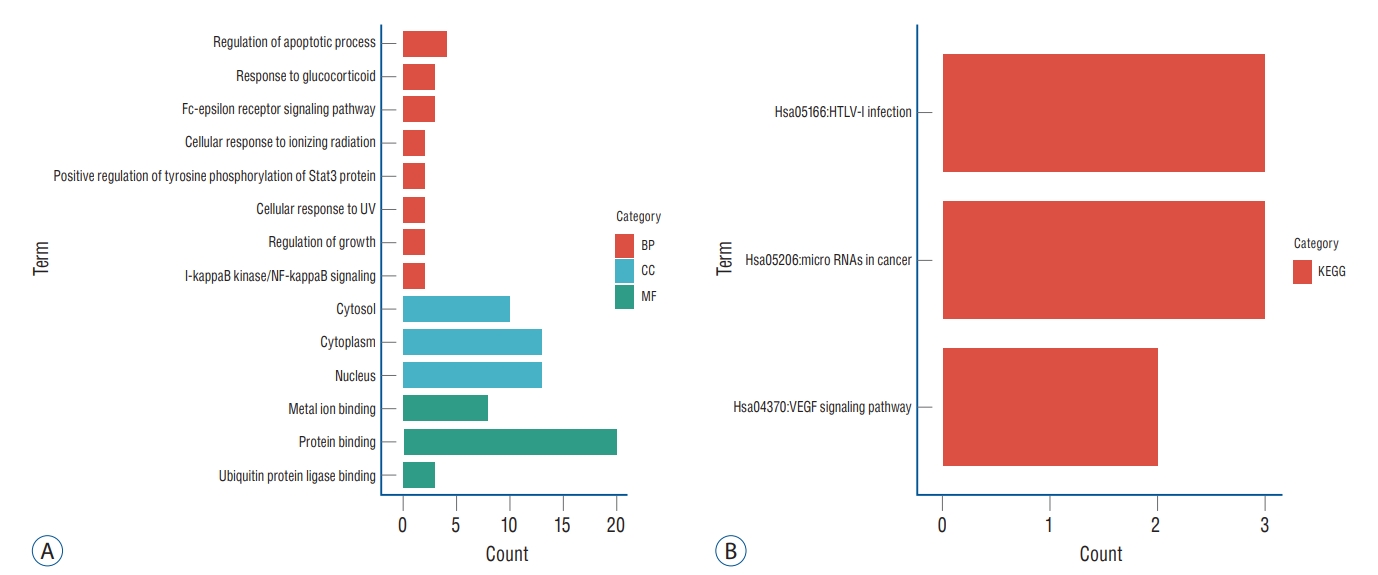
GO and KEGG analyses of the 27 up-regulated genes and nine down-regulated genes were performed using DAVID. Finally, a total of 14 GO items with statistically significant were obtained, including three CC entries, eight BP entries, and three MF entries. Results showed that GO BP was significantly enriched in Regulation of apoptotic process, Response to glucocorticoid, Fc-epsilon receptor signaling pathway and other processes; CC was mainly in cytoplasm and Nucleus processes; and MF was mainly enriched in protein binding process. The above-mentioned results were shown in Fig. 5 and Table 2. Enrichment analysis of KEGG pathway showed that DEGs was significantly enriched in HTLV-I infection and miRNAs in cancer, the results of functional analysis were shown in Fig. 5 and Table 3.
Construction of PPI network and selection of hub gene
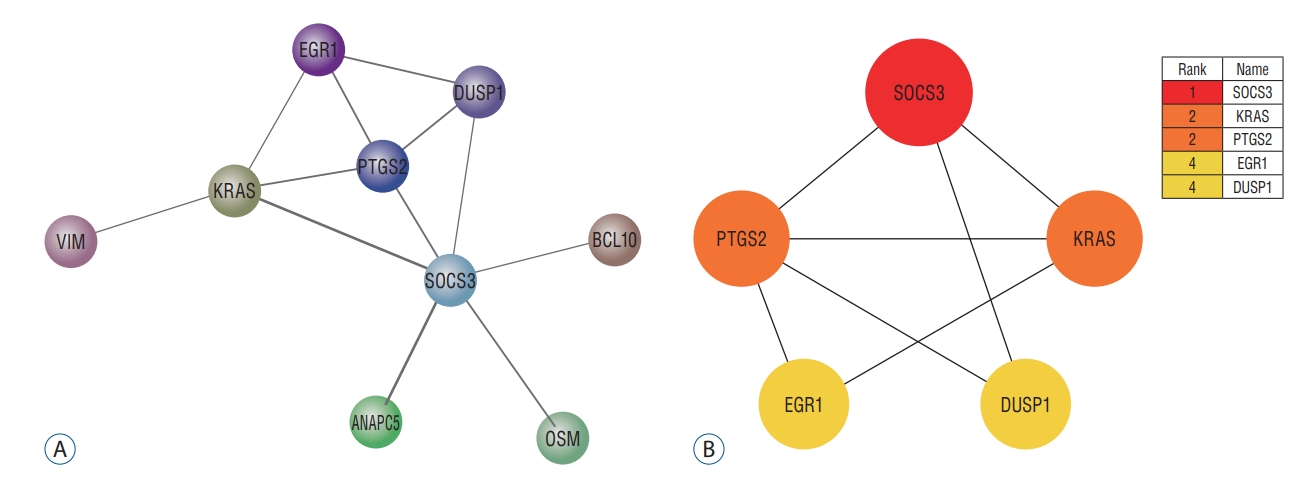
The PPI network of DEGs was constructed using STRING ver. 10 and the result was shown in Fig. 6A. And then, the PPI network of the 27 up-regulated genes and nine down-regulated genes was analyzed by Cytoscape software. The PPI information was analyzed by cytohubba software for determining the core targets in the network. Eventually, five core genes were obtained, namely SOCS3, KRAS, PTGS2, EGR1, and DUSP1. According to the node degree score generated by Cytoscape, the potential hub genes were identified, including SOCS3 (6 points), KRAS (4 points), PTGS2 (4 points), EGR1 (3 points), and DUSP1 (3 points). The results were shown in Fig. 6B.
Integrated miRNA/gene regulatory network
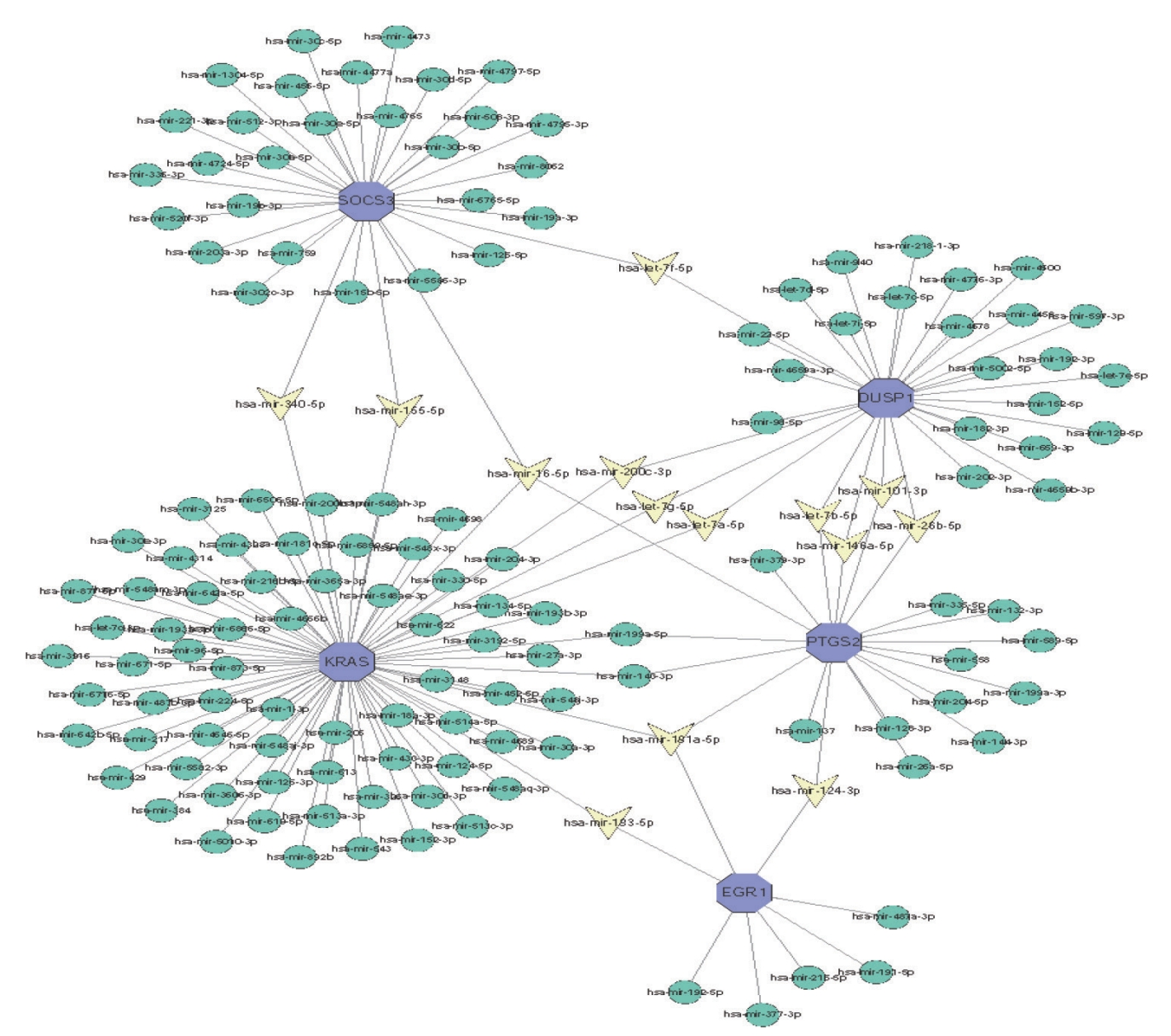
The significantly different miRNA gene regulatory network was constructed by using Cytoscape software, and miRNA was predicted based on the network analysis database. The top 5 DEGs and their corresponding regulatory miRNAs molecules were shown in Fig. 7. The results showed that KRAS, PTGS2, and SOCS3 could be used as the common target for predicting hsa-mir-16-5p, whereas the common targets of hsamir-181a-5p were KRAS, PTGS2, and EGR1, the common targets of hsa-mir-124-3p were PTGS2 and EGR1. The above findings were needed to further validate in future research.
DISCUSSION
In the present study, the gene chip information of IS was obtained through GEO database, and a total of 36 DEGs, containing 27 up-regulated, and nine down-regulated DEGs. Then, GO function analysis and KEGG pathway enrichment analysis of DEGs were performed, and the results of GO analysis were closely related to regulation of patriotic process, cytoplasm and protein binding, while the results of KEGG analysis were mainly related to HTLV-I infection and miRNAs in cancer pathway. To analyze the interaction between DEGs genes, PPI network diagram was constructed, and the results indicated that SOCS3, KRAS, PTGS2, EGR1, and DUSP1 genes are closely related to IS pathological process. In addition, gene regulatory network had demonstrated that multiple genes could be used as the targets of hsa-mirRNA.
Five core genes related to IS were identified from PPI network diagram. Suppressor of cytokine signaling (SOCS) plays an important regulatory role in the process of cytokine signal transduction. SOCS is ubiquitous in various tissues and organs, with a total of seven superfamilies, among which SOCS3 is related to the signal regulation function of tumor necrosis factor-╬▒ (TNF-╬▒) [26]. SOCS3, an inhibitory regulator mediated by cytokines, is closely related to the occurrence and negative regulation of inflammatory reaction. TNF-╬▒, one of the important inflammatory cytokines that induce SOCS3 activation, can regulate SOCS3 gene by inducing Janus kinase/signal transducers and activators of transcription 3 signal pathway, thereby up-regulating the expression of SOCS3 gene and preventing the degradation of SOCS3 protein [8,12,24]. Moreover, SOCS3 happens to be an important negative regulator in aforementioned signaling pathway, which may lead to the inhibited the activity of Janus kinase/signal transducers and activators of transcription 3 signaling pathway [34]. Previous studies have demonstrated an association between the IS abnormal activation of immunity and the occurrence and development of IS, which might result in abnormal levels of inflammatory factors in vivo [38]. Studies have shown that the more severe the condition of IS patients, the more severe the inflammatory reaction in the body and the more expression of inflammatory factors, which not only promotes the formation, development, rupture and thrombosis of atherosclerotic plaques, but also participates in the early reperfusion injury and inflammatory reaction in patients with IS [4]. SOCS3 is related to inflammation, and inflammation plays a key role in the occurrence and development of IS. Therefore, we speculated that SOCS3 may participate in the pathogenesis of IS through inflammatory factors. The PTGS2 gene is located at 1q25.2-q25.3 has a total length of 813 kb, including 10 exons and nine introns, and encodes COX-2. The expression of PTGS2 gene is highly restricted under basic conditions and is almost not expressed under normal physiological conditions. However, under many pathological conditions, COX-2 may overexpress due to various stimuli from the internal and external environment.
Prostaglandins (PGs), produced by COX-2 catalyzing arachidonic acid, is one of two important molecules that mediate inflammation. The expression of COX-2 and the synthesis of PGs play an important role in the pathophysiological process of chronic inflammation. Overexpression of COX-2 may lead to inflammation of blood vessel walls, plaque instability and intimal hyperplasia [32]. Therefore, the cyclooxygenase gene PTGS2 encoding COX-2 may be closely related to the occurrence of stroke through inflammation, and the PTGS2 gene polymorphism has been confirmed to be associated with the occurrence of stroke. A small case-control study found that the G765C polymorphism of PTGS2 gene was positively correlated with the occurrence of ischemic cerebrovascular disease, and remained positive correlation after adjusting for traditional risk factors [5]. Kohsaka et al. [17] found that the G765C polymorphism of the PTGS2 gene was a risk factor for stroke and coronary heart disease. Therefore, the results of our study indicated that the correlation between the PTGS2 gene and IS was consistent with the results of previous studies, but the specific mechanism remained to be further studied.
Early growth response 1 (EGR1), also known as nerve growth factor inducible factor, is a member of the immediate early genes, which is characterized by rapid and short-time activation after a series of external stimuli. EGR1 is hardly expressed or low-expressed in normal somatic cells. Once cells are stimulated by a variety of extracellular signals, the body can activate the transcription of EGR1 through one or more signal transduction pathways. Activated EGR1 can regulate cell function and exert various biological effects after binding to specific binding sites on target gene promoters. As an exogenous stimulus, ischemia and hypoxia can induce cells to rapidly overexpress EGR1. As an important nuclear transcription factor, EGR1 plays an important role in regulating cell growth, differentiation, development, proliferation, and inflammatory response. Especially in the inflammatory response, EGR1 can initiate and amplify the inflammatory response. In the brain tissue after IS, the post-stroke inflammatory response induced by overexpression of EGR1 may cause secondary damage to the ischemic brain tissue [25]. Furthermore, EGR1 is involved in a variety of pathophysiological processes, including cell proliferation, differentiation and apoptosis, thrombosis, inflammation, and DNA repair [9,25,28,30]. It has been reported that EGR1 could aggravate inflammation and nerve injury after ischemia [33]. The results of the present study indicated that SOCS3, PTGS2, and EGR1 were the core genes for the occurrence of IS, most of which were related to inflammation. Inflammation is the core link of IS damage and is closely related to the occurrence of IS. We therefore speculated that hub genes may participate in the occurrence of IS by affecting the inflammatory response. This finding was in accordance with those of previous studies, however, the underlying mechanism in detail required further study.
The five hub genes in the pathogenesis of IS were identified by using the STRING software of Cytoscape. The results of miRNA regulation showed that multiple hub genes could be used for predicting the common target miRNA, and miRNA could regulate gene expression by combining with the 3ŌĆÖUTR of the target gene. With the continuous deepening of research, the regulatory scope and diversity of miRNA regulatory molecules have been confirmed by increasing number of studies [15,37]. MiRNA not only involves the normal BP of the host, during which it inhibits the transcription of the target gene and regulates the cell cycle, cell differentiation, apoptosis, angiogenesis and metabolism, but also participates in the regulation of oxidative stress, inflammation of the body, cell proliferation and migration [20,39]. There is a certain correlation between miRNA and stroke. During the onset of stroke, some miRNAs can play the role of anti-ischemia, anti-oxidation, and protection of cranial nerves, while some miRNAs are related to reperfusion nerve damage [36]. It was found that the levels of serum miR-124, miR-134 and inflammatory factors were abnormally increased, and miR-124 and miR-134 were related to the severity of the disease and inflammatory reaction. Detection of serum miR-124 and miR-134 may be an important means for clinical diagnosis of stroke and assessment of the disease [10]. Considerable studies have shown that miRNAs play an important role in the occurrence and development of various cardiovascular diseases, and miRNAs are involved in the process of apoptosis in cardiovascular diseases [22], and thus miRNAs may be potential targets for the treatment of cardiovascular diseases. In our study, there were three miRNAs closely related to IS, including hsa-mir-16-5p, hsa-mir-181a-5p and hsa-mir-124-3p, all of which had been reported in diseases. Mir-16-5p can regulate thioredoxin-interacting protein (TXNIP), while TXNIP can regulate the redox state of cells and induce oxidative stress, resulting in inflammation or apoptosis. In addition, mir-16-5p can affect lipopolysaccharide-induced cardiomyocyte damage through TXNIP [1].
It has been reported that mir-16 can regulate the apoptosis of lung epithelial cells after oxidative stress [2], which indicated that mir-16 may be involved in oxidative stress. In the present study, mir-16-5p might play a role in the occurrence of IS by regulating oxidative stress. Mir-124 -3p is of great significance in regulating oxidative stress and inflammatory response induced by traumatic brain injection (TBI). The expression of mir-124-3p is down-regulated after TBI. Therefore, up-regulation of mir-124-3p can inhibit the release of inflammatory factors, and overexpression of mir-124-3p can target and regulate the expression of downstream proteins, inhibit the inflammatory reaction of neurons and alleviate the development of TBI [11]. Mir-181a-5p is up-regulated in various diseases such as vasculitis and sepsis, and can promote the occurrence of inflammatory reactions [6]. Mir-181a-5p has been proved to be involved in the occurrence and development of various tumors. Relevant studies on liver cancer found that mir-181a-5p has the function of tumor suppressor gene, and its expression in liver cancer tissue is reduced or even absent, which inhibits the function of tyrosine protein kinase receptor, thereby inhibiting the growth of tumor [18]. Mir-181a-5p is also considered as tumor suppressor gene in breast cancer, which has a significant inhibitory effect on breast cancer cell proliferation and promotes breast cancer cell apoptosis after transfection [21]. Chen et al. [3] found that gastric cancer tissue showed high expression of mir-181a-5p, which was positively correlated with the invasion and metastasis of gastric cancer. Meanwhile, the expression of mir-181a-5p in vitro was up-regulated, which could promote cell proliferation and metastasis, and has an effect similar to cancer-promoting genes [3]. In our study, the specific role of hsa-mir-16-5p, hsa-mir-181a-5p and hsa-mir-124-3p remained unclear, but they might be related to oxidative stress and inflammatory reaction, thus participating in the progress of IS.
In view of the correlation between Hub gene and IS, as well as the correlation between miRNA and IS in this study, it can be known that the occurrence of IS was related to oxidative stress and inflammatory reaction. Several evidences have shown that the sudden decrease or interruption of glucose and oxygen supply leads to the rapid disorder of brain energy metabolism, which cause the generation of a large number of reactive oxygen species and oxidation intermediates, resulting in severe oxidative stress injury in a short time. Oxidative stress is the result of injury process and self-regulation mechanism under the imbalance of oxidation and antioxidation, and its core lies in the production of reactive oxygen species. Inflammation is one of the major sources of reactive oxygen species in subacute stroke. A large amount of reactive oxygen species produced in acute stroke can induce early inflammation, and then lead to the activation of various immune cells. There is also a relationship between oxidative stress and inflammation. Oxidative stress can induce the damage of cell structure and the appearance of inflammatory reaction, and result in cell death after stroke in many ways [7]. The above-mentioned evidences indicate that bioinformation analysis results are consistent with the laboratory results.
This study obtained information from the database for bioinformatics analysis to find the key genes of IS, it also revealed that inflammatory response and oxidative stress are closely related to the pathophysiological process of IS. Targeted therapy of key pathogenic genes such as SOCS3, KRAS, PTGS2, EGR1, and DUSP1, may provide new ideas for the treatment of IS.
Bioinformatics analysis plays an important role in the treatment of stroke, which contributes to better understand of the stroke pathogenesis and provide effective and novel strategies for the treatment. Conversely, the study has certain limitations : 1) the small number of IS patients and normal group patients in the two microarray data, may affect the final results, and more samples are needed for verification, furthermore bioinformatics obtained with small samples can bias the results, and the actual bioinformatics result is different due to data change; 2) the specific molecular mechanism of hub genes and miRNAs in stroke regulation is insufficient; 3) in the constructed network, there is a lack of research on the functions of hub genes and miRNA; 4) this study is mainly bioinformatics analysis, and some changes in the tools used in the analysis may lead to different results; and 5) bioinformatics obtained with small samples can bias the results, and the actual bioinformatics result is different due to data change.
CONCLUSION
Taken together, the present study identified DEGs related to IS using bioinformatics theories and techniques. These DEGs participated in BPs such as regulation of patriotic process, cytoplasm, protein binding, etc., and mediated signal pathways such as HTLV-I infection and miRNAs in cancer. Result of PPI indicated that DEGs were related to mediate the occurrence and development of stroke. SOCS3, KRAS, PTGS2, EGR1, and DUSP1, as Hub genes of stroke, were closely related to the pathological process of disease occurrence. Most of the top genes were related to inflammatory factors, which might participate in the occurrence of stroke through inflammatory reaction. The results of miRNA gene regulation confirmed that hsa-mir-16-5p, hsa-mir-181a-5p and hsa-mir-124-3p might be key miRNAs, and all three miRNAs were related to oxidative stress and inflammation. Therefore, it was speculated that three core miRNAs may influence the mechanism of stroke through oxidative stress and inflammation. The identification of genes and miRNAs mentioned above may contribute to the development of early diagnosis strategies, prognostic markers and therapeutic targets of stroke. Nevertheless, further experimental studies are still required to verify the functions of these molecules in stroke.