INTRODUCTION
Craniosynostosis is premature fusion of one or more cranial sutures, which is usually encountered early in life. This relatively common disorder of the skull affects between 1 in 2100 to 2500 children1,11). Pediatricians are usually the first physicians who encounter the craniosynostosis patients. Most cases are sporadic, but may have an underlying genetic basis. Skull deformities have a wide range of etiologies from positional plagiocephaly to complex genetic syndromes. Craniosynostosis can be divided into primary, secondary, syndromic or nonsyndromic; more than 85% of patients have nonsyndromic craniosynostosis2). Primary single-suture synostosis is relatively easy to diagnose because the main symptom is skull deformity, which is typical according to the fused suture. Multiple suture synostosis is less common than single suture, accounting for ~15% of cases. The intracranial pressure is usually normal in primary craniosynostosis, although ~20% of single-suture synostosis patients have high intracranial pressure18,21). Brain damage in cases of single-suture synostosis is usually mild but requires prolonged monitoring7). Increased intracranial pressure and brain damage are more common in secondary or syndromic craniosynostosis. Secondary or syndromic craniosynostosis is more difficult to diagnose despite recent radiological advancements. Syndromic craniosynostosis should be diagnosed as early as possible to facilitate interdisciplinary management, improve outcomes and provide genetic counseling for parents. In this regard, the authors reviewed diagnostic evaluation with a focus on pediatric systematic evaluation and genetic studies.
OVERVIEW OF SECONDARY AND SYNDROMIC CRANIOSYNOSTOSIS
Secondary craniosynostosis results from a known underlying disorder, which can include systemic and metabolic conditions such as hyperthyroidism, hypercalcemia, hypophosphatasia, vitamin D deficiency, renal osteodystrophy, Hurler's Syndrome, sickle cell disease, thalassemia, and encephalocele10,19). Secondary craniosynostosis also develops in cases with post-ventriculoperitoneal shunt and microcephaly due to developmental failure of the brain. Syndromic craniosynostosis is suspected when a patient has psychomotor retardation, digital anomalies, skeletal defects, cardiac defect, or other organ anomalies; most are caused by genetic variants. To date, various causal genes and over 180 syndromes have been reported15).
Secondary or syndromic craniosynostosis needs a more systematic approach. It is important not only to distinguish craniosynostosis from positional skull dysmorphism but also to evaluate the cause of the condition. Next-generation sequencing (NGS) technology gives hope to a large number of patients with genetic craniosynostosis by sequencing many candidate genes simultaneously at a reasonable price. In this review, we will focus on the diagnostic evaluation of syndromic craniosynostosis including NGS-based genetic analysis.
SYSTEMATIC EVALUATION OF CRANIOSYNOSTOSIS
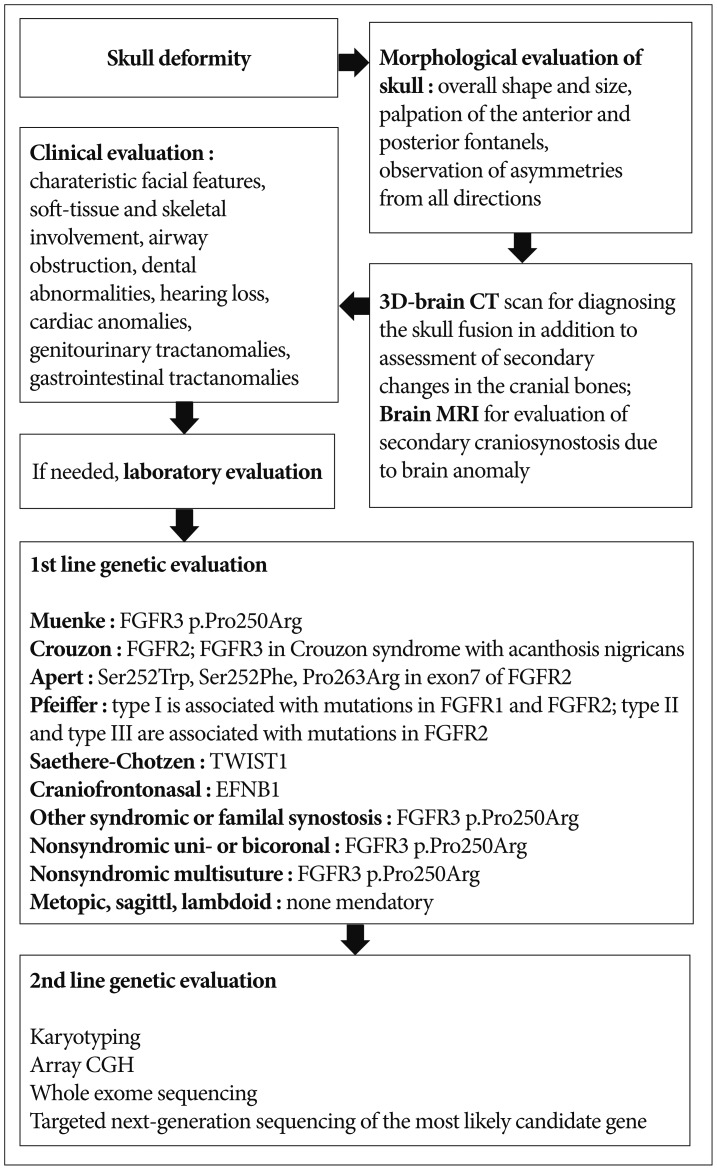
Systematic evaluation includes morphological, imaging, clinical and laboratory tests. Morphological evaluation of the skull should be performed for overall shape and size, and palpation of the anterior and posterior fontanels with attention to size, shape, and fullness with the infant in both the upright and supine positions. In addition to looking at the infant from the front and sides, it is important to observe the skull shape from above, particularly to note any asymmetries in ear position and any flattening of the skull posteriorly, as well as from behind so the levelness of the skull base can be assessed. Deformational plagiocephaly resulting from external molding forces is one of the common causes of cranial asymmetry. Risk factors of positional plagiocephaly are male sex, prematurity, and torticollis. Causes of torticollis include cervical spine abnormality, infection, congenital muscular torticollis, etc. Congenital muscular torticollis can be diagnosed by ultrasonography of the sternocleidomastoid muscle. The most common deformational skull shape is occipital asymmetry. Positional deformation is different from craniosynostosis in that the parallel quadrangular shape results from a positional effect instead of trapezoid shape and compensatory contralateral bulging in craniosynostosis. Occipital flattening is a relatively common feature in oriental neonates and should not be confused with bilateral lambdoid synostosis. The incidence has increased in western countries since the recommendation of putting babies to sleep on their back to prevent sudden infant death syndrome. Sometimes, multiple synostotic patients present with a symmetric, normal-looking appearance. In this case, the skull is small for their age. Close follow up of head circumference and developmental assessment are important in suspected patients. Most cases of craniosynostosis can be diagnosed by their abnormal morphologies and routine radiological examination is not needed. The mainstay of craniosynostosis imaging is three-dimensional CT scan, which facilitates diagnosis of skull fusion in addition to assessment of secondary changes in the cranial fossae, orbits and facial bones. It also enables differentiation of craniosynostosis and positional plagiocephaly. Radioisotope bone scan is helpful; however, it is not used in the clinical setting. Brain magnetic resonance imaging (MRI) is useful for evaluating secondary craniosynostosis due to brain anomalies. If secondary or syndromic craniosynostosis is suspected, further diagnostic procedures are needed. Details of radiologic evaluation will not be discussed in this review. Clinical evaluation is important to assess additional features suggesting a syndrome or complications that need urgent management. Coronal synostosis is the most common type of craniosynostosis associated with other anomalies, suggesting a syndromic nature20). The presence of characteristic facial features and malformations is important for diagnosis of a syndromic craniosynostosis. Special attention should also be paid to the functional consequences of the condition. Decreased level of consciousness, neurologic deficits, breathing difficulty, choking or vomiting on feeds, and irritability may be indications for acute intervention8). Abnormalities of extremities are diagnostic for syndromes; for example, a broad medially deviated thumb or big toe in Pfeiffer syndrome, and more extensive syndactyly in Apert syndrome. Craniosynostosis involving multiple sutures frequently extends to premature fusion of the skull base and shows varying degrees of midfacial underdevelopment, shallow orbit, exophthalmos, low-set ears, narrow, highly arched palate, and malocclusion20). Soft-tissue and skeletal involvement, airway obstruction, dental abnormalities, hearing loss, cardiac anomalies, genitourinary tract anomalies, and gastrointestinal tract anomalies may also be associated and should be assessed in syndromic craniosynostosis5,1419). Laboratory evaluation of thyroid hormone, calcium, phosphate, alkaline phosphatase, and vitamin D levels can serve as adjuncts to history and examination. A flow chart of clinical and genetic diagnosis of craniosynostosis is shown in Fig. 1.
GENETIC EVALUATION OF CRANIOSYNOSTOSIS
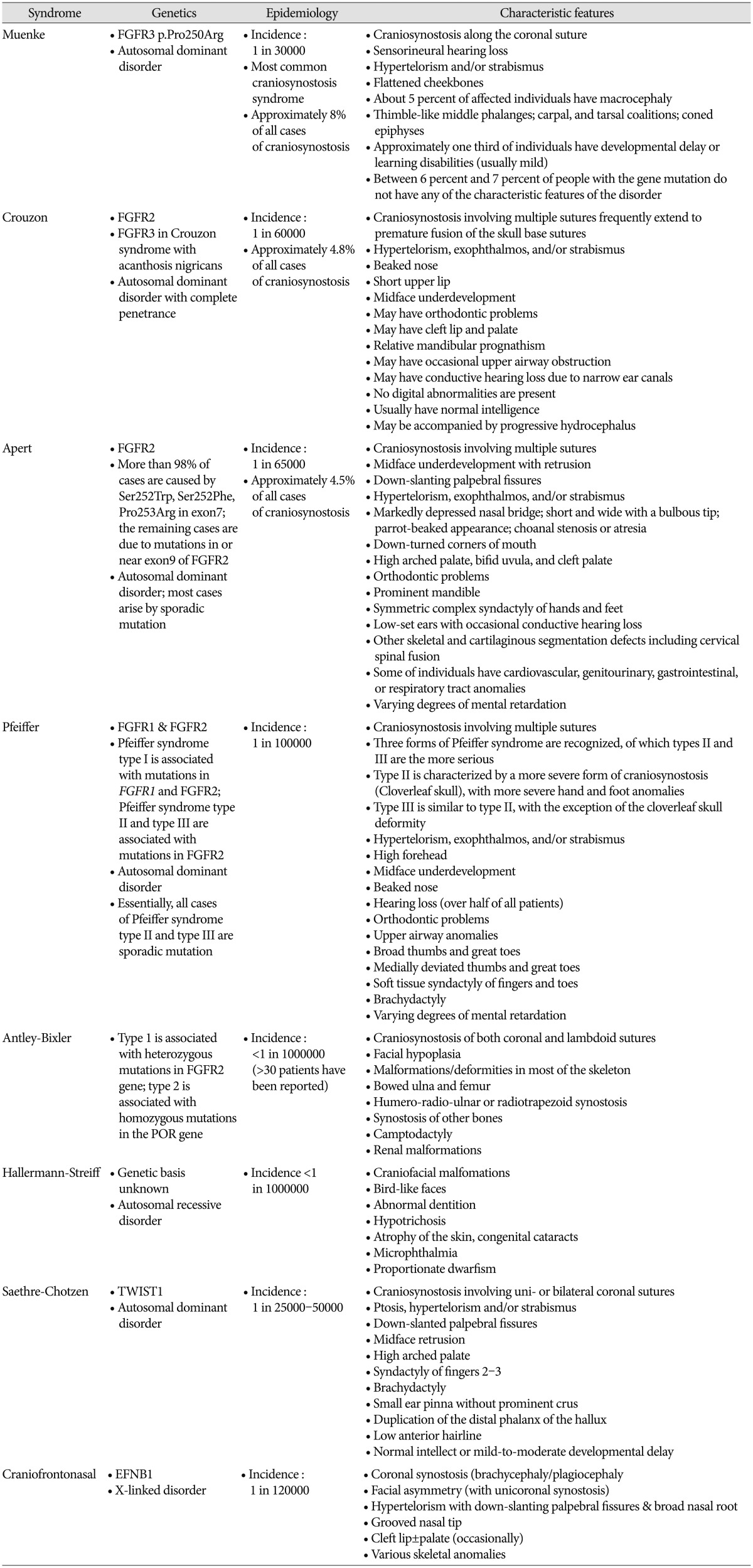
Molecular genetic evaluation of craniosynostosis is important because the results can provide information about the etiology of the disease and predict the clinical course and prognosis of the patient6). About 45% of unselected cohorts of craniosynostosis patients have a causal genetic alteration detected by current genetic testing strategies6,22). When selecting candidates for molecular genetic testing, various factors-including involved sutures, associated anomalies, developmental milestones, and family history-should be considered. Karyotyping and array comparative genomic hybridization (CGH) are recommended as basic molecular genetic tests for these patients6). Various chromosomal aberrations have been associated with syndromic craniosynostosis, and account for at least 10% of the cases3,416). With the increasing availability and use of array CGH, copy number variants (CNVs) such as submicroscopic deletions and duplications have emerged as important causes of craniosynostosis. Mefford et al.12) reported that 7.5% of individuals with single-suture synostosis have at least one rare CNV. Trigonocephaly also results from genetic abnormalities, such as deletion of chromosome 9p22-p24 and 11q23 (Jacobsen syndrome)20). Some patients with 22q11.21 microdeletion show craniosynostosis8). Other CNVs-including 1q43, 2p21, 2q14, 3p25, 5p15, 6p21, 6q26, 7q36, 9q21, 11q25, 12p21, and 17q25-have been identified in affected individuals12). At the gene level, most abnormalities related to craniosynostosis are FGFR-, TWIST1-, and EFNB1- related syndromes16). The genetics, epidemiology and characteristic features of syndromic craniosynostosis are summarized in Table 1. According to Wilkie et al.22)
FGFR2, FGFR3, TWIST1, and EFNB1 account for ~25% of craniosynostosis. Much rarer genetic mutations are in FGFR1 (mild Pfeiffer syndrome), POR (Antley-Bixler syndrome), RAB23 (Carpenter syndrome), EFNA4 (non-syndromic coronal synostosis)13), ESCO2 (Roberts syndrome), GLI3 (Greig syndrome), JAG1 (Alagille syndrome), KRAS (Noonan syndrome), RECQL4 (Baller Gerold syndrome), TGFBR1 or TGFBR2 (Loeys-Dietz syndrome), and MSX2 (single family with supraorbital recession; several duplications have been associated with craniosynostosis)9). L1CAM is associated with development of white matter and its mutation can manifest callosal agenesis, mental retardation, adducted thumbs, spasticity, or hydrocephalus15). A defect in the interaction of FGFR with L1CAM may be the cause of the brain malformations and mental retardation in patients with craniosynostosis17). Genetic consultation based on the result of the evaluation is an important role of the physician responsible for the patient. When family history is negative, a sibling recurrence risk is 2% for metopic, sagittal and lambdoid synostosis, 5% for unicoronal synostosis and 10% for bicoronal and multisuture synostosis8). Genetic evaluation is not mandatory for metopic, sagittal or lambdoid synostosis because the recurrence risk is low and they seldom cause complications. When there is a family history, offspring risks are ~5% in the case of nonsyndromic sagittal, metopic and unicoronal synostosis; 30-50% in bicoronal and multisuture synostosis8). Targeted genetic testing should be performed for patients in whom a specific diagnosis is suspected. If a wide range of mutations is found in the suspected causal gene or multiple candidate genes are present, whole-exome sequencing or targeted next-generation sequencing is faster and less costly than Sanger sequencing. Genetic testing for the FGFR3 p.Pro250Arg mutation should be offered for all patients presenting with coronal or multisuture synostosis because it is the most commonly found mutation in all cases of craniosynostosis8). If no genetic abnormalities are identified in the first-line genetic evaluation, the second-line evaluation should include karyotyping and array CGH. Whole-exome sequencing or targeted next-generation sequencing should than be considered if not performed in the first-line genetic evaluation.