Sellar-Suprasellar Extraventricular Choroid Plexus Papilloma : A Case Report and Review of the Literature
Article information
Abstract
Choroid plexus papillomas (CPPs) are relatively rare neuroectodermal tumors that develop from choroid plexus epithelial cells and are usually restricted to the ventricles. Extraventricular CPPs are very unusual and can be difficult to diagnose and treat. A 50-year-old male patient was admitted to our clinic complaining of headache and visual deterioration. Neurological examination found no abnormalities except decreased light perception and secondary optic atrophy in the left eye. Endocrine testing revealed normal levels of hormones produced by the pituitary and target glands. Magnetic resonance imaging of the brain revealed a huge regular-shaped lesion in the sellar-suprasellar region occupying the sella turcica and extending into the suprasellar cistern and planum sphenoidale. The lesion was completely excised by microsurgery via an ordinary left-sided pterional approach. Histopathology identified the lesion as a choroid plexus papilloma. Following the case report, literature on the origin, differential diagnosis, and treatment of this rare tumor is reviewed.
INTRODUCTION
Choroid plexus papillomas (CPPs) are relatively rare neuroectodermal tumors that develop from choroid plexus epithelial cells and account for 0.4–0.6% of all primary brain tumors7). CPPs are usually restricted within the trigone of the lateral and fourth ventricles and have clear borders7). In adults, CPPs generally develop in the fourth ventricle, but in rare cases they can occur in extraventricular sites such as the cerebral parenchyma7), cerebellopontine angle1213), or suprasellar region917). The exact mechanism by which CPPs arise at extraventricular sites remains unclear and is subject to some controversy. In this report we present an unusual case of sellar-suprasellar CPP and review the literature.
CASE REPORT
A-50-year-old male patient was admitted to our clinic with headache and a 6-month history of progressive deterioration of vision in the left eye. Vision from the right eye was normal and no bitemporal hemianopsia or symptoms of endocrine disturbance were observed. He had also suffered headache for the last month. The only abnormal findings on neurological were decreased perception of light and secondary optic atrophy in the left eye. Endocrine testing revealed normal levels of hormones produced by the pituitary and target glands.
Magnetic resonance imaging (MRI) of the brain revealed a huge regular-shaped lesion in the sellar-suprasellar region occupying the sella turcica and extending into the suprasellar cistern and planum sphenoidale. On T1-weighted images, the majority of the mass was of low intensity, suggesting a cystic lesion. The right side base of the lesion, just above the suprasellar carotid artery, contained a hyperintense contrast-enhanced nodule. On T2-weighted images, the lesion was hyperintense, also compatible with a cystic lesion. The suprasellar cistern was filled and the optic chiasm and third ventricle were elevated by the lesion (Fig. 1). According to these findings, the lesion was diagnosed as a craniopharyngioma, pituitary adenoma, cystic astrocytoma or ependymoma.

Radiological findings. A : T1-weighted axial MRI reveals a hypointense regular-shaped cystic lesion at the ventricular border. B : Contrast-enhanced T1-weighted coronal MRI reveals that the lesion compresses the optic chiasma and elevates the third ventricle and identifies the contrast-enhanced solid hyperintense nodule at the right floor of the lesion, just above the suprasellar internal carotid artery. C : Hyperintense cystic lesion on T2-weighted axial MRI. D : Tumoral invasion of the planum sphenoidale, whole sellar area, and suprasellar cistern.
The tumor was approached via a left pterional craniotomy. The cystic component of the tumor was reached first and upon opening the cyst wall, the contents were aspirated. On the basal right side of the lesion, a yellowish vascular nodule adjacent to the suprasellar internal carotid artery was revealed (Fig. 2) and completely excised while preserving the internal carotid artery. During the operation, no communication with the third or lateral ventricles was apparent.
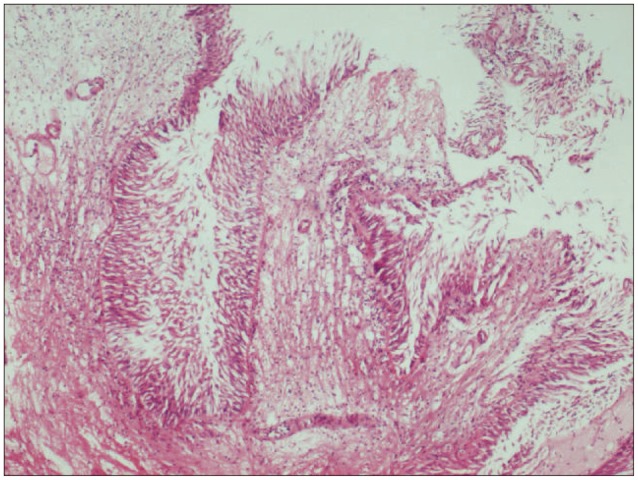
Histology reveals the tumor's papillary structure with one or more layers of columnar epithelial cells around a fibrovascular core (H&E, ×40).
Histopathological investigation revealed papillary structures with a delicate fibrovascular core lined by one or more layers of columnar epithelial cells. Immunohistochemistry found that tumor cells expressed pancytokeratin (pan-CK), but not glial fibrillary acidic protein (GFAP). The tumor was thus diagnosed as CPP (Fig. 3, 4). Following surgery, the patient's vision in his left eye improved rapidly and he reported no adverse events or changes in neurological function. Postoperative MRI showed total excision of the tumor (Fig. 5). The patient is currently under outpatient observation.

Micrograph of immunostained section shows positive expression of pancytokeratin (×10).

T1-weighted contrast enhanced axial (A) and T2-weighted sagittal (B) postoperative MRI show total excision of the tumor.
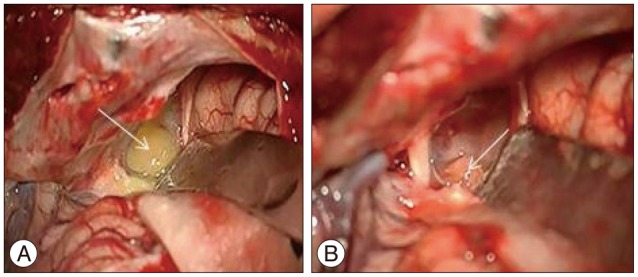
Intraoperative image. A : The cystic component of the tumor (white arrow). B : Yellowish tumoral nodule (white arrow) after opening the cystic component.
DISCUSSION
Choroid plexus papillomas are rare tumors of the central nervous system, representing less than 1% of all verified intracranial neoplasms11). These neoplasms are commonly confined to the ventricle system, where the choroid plexus is normally located; in the lateral ventricles in infants and children and in the fourth ventricle in adults11). A few reports have described CPPs arising from extraventricular sites such as the posterior third ventricle1415), cerebellopontine angle1213), posterior fossa36), brain stem16), sacral canal10), and cerebral parenchyma7).
CPPs rarely occur in the pituitary fossa and sellar/suprasellar region. To our knowledge, only five cases have been reported in the English-language literature, including our case491117).
In Table 1 we summarize the important features of previously reported cases. The case reported by Winer et al.18) was excluded from this table because of the possibility that the tumor arose in the third ventricle and extended downwards into the sella.

Summary of previously reported cases
The exact mechanism by which CPPs arise in extraventricular sites remains unclear and is subject to some controversy. Most extraventricular CPPs are located at the cerebellopontine angle (CPA). Tumors in this location can result from herniation of the tumor through the foramen of Luschka or from de novo development in the choroid plexus lying outside the fourth ventricle at the CPA, referred to as Bochdaleck's flower basket48).
Two hypotheses have been suggested for the origins of extraventricular intraparenchymal CPPs by Azzam and Timperley1) : first, that they might arise from primitive ectopic choroid plexus in the extraventricular site and second, that they may develop from epithelial tissue that migrated to extraventricular areas during brain development. In our case, neuroimaging and operative findings clearly showed that the CPP was not attached to the ventricular choroid plexus and had not metastasized from another CPP. Therefore, we infer that the CPP of the sellar region in our case arose from ectopic choroid plexus tissue as in the cases reported by Bian et al.4), Ma et al.11), and Sameshima et al.17).
Imaging characteristics were not sufficiently distinct to preoperatively diagnose CPP in our case, similar to other reports41117). CPP typically appears on CT as a well-defined, homogeneous enhancing mass with lobulations and a frond-like irregular pattern, resulting in a cauliflower-like appearance10).
Other reported cases491117) appeared nearly identical; however, our case had completely different neuroimaging characteristics, including parasellar cystic extension and a hyperintense contrast-enhanced nodule. Overall, distinguishing a CPP from a pituitary adenoma or other pathologies is difficult based only on neuroimaging.
Complete microsurgical excision of the tumor is the recommended therapy and was achieved in our case using a left-sided pterional approach5). In other cases the most common neurosurgical procedure is an endonasal transsphenoidal approach411)17). The only report of excision via a pterional approach similar to ours is from Kimura et al.9). We chose the pterional approach for two reasons : first, the tumor had marked supra- and parasellar extensions and second, it seemed the only safe means of avoiding the suprasellar internal carotid artery. Except for undifferentiated forms of the tumor, CPP does not metastasize through the cerebrospinal fluid2), so postoperative radiotherapy was not considered in our case, similar to previous reports11).