INTRODUCTION
Neuroblastic tumors present a wide spectrum of tumors, including the benign ganglioneuroma, intermediate ganglioneuroblastoma and undifferentiated neuroblastoma (NB)38). Ganglioneuromas are benign tumors, which usually arise from the thoracic cavity as mediastinal tumors or abdominal cavity as retroperitoneal mass lesions4,25,30). Ganglioneuroblastomas are tumors with decreased differentiation, which consist of a ganlioneuromatous stroma and a neuroblastomatous component38). NB is a common malignant tumor in pediatric age group, on contrary to that it is a rare entity in adults 11,19). NB represents 8-10% of all childhood cancers. Spinal involvement is rare and spinal NB in adulthood is extremely rare2,7,18,37). We report a case of spinal NB recurrent from a ganglioneuroblastoma after disease free survival of 13 years. To our knowledge, it is the second spinal case in the literature indicating such a malignant transformation and the only case with further genetic investigations.
CASE REPORT
A 22-year-old male patient admitted with abdominal pain to the general surgery department. His physical examination was normal. An abdominal computed tomography scan was obtained and a retroperitoneal mass lesion with spinal involvement was observed. The patient was then referred to neurosurgery department. His neurological examination revealed no abnormalities. Magnetic resonance imaging (MRI) of the thoracolumbar spine showed a retroperitoneal mass lesion at T12-L3 level, extending from the left paraspinal region into the spinal canal. Using a combined lateral and posterior approach, T12-L3 laminectomy and left L1-2 facetectomy was performed. The tumor was near totally removed with only a small retroperitoneal remnant, which could not be reached with the current approach. Pathological diagnosis was ganglioneuroma. Postoperative course was uneventful. The patient was discharged from the hospital with no neurological deficits.
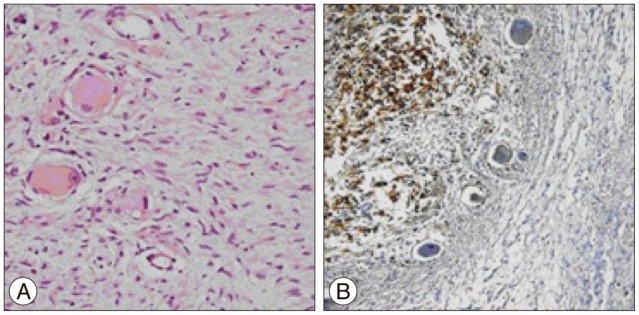
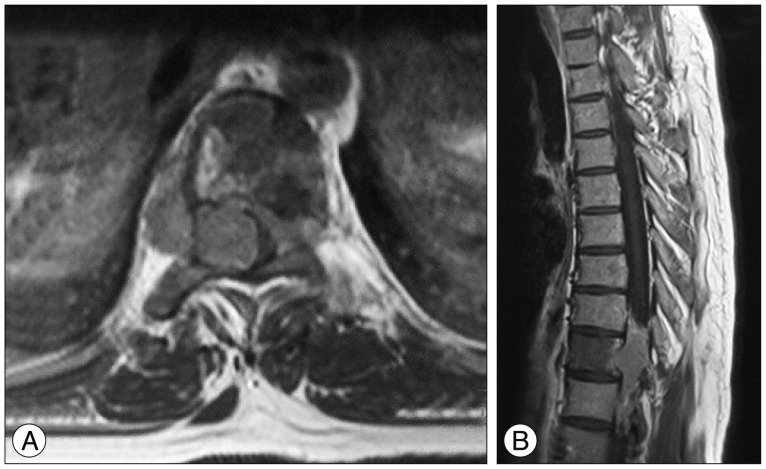
After four years with stable disease, the patient presented with the same complaints. His neurological examination was normal. MRI revealed a T11-L3 left paraspinal mass growing towards T11-12 neural foramen and spinal canal (Fig. 1). With the same approach tumor was subtotally removed (except the retroperitoneal extension) and spinal cord was decompressed. Macroscopic examination revealed a grey coloured, lobulated, soft tumor with a capsule and 8├Ś7.5├Ś5 cm in size. The cut surface was yellow-white coloured and included hemorrhagic areas. Histopathological diagnosis was ganglioneuroblastoma. While there was a inconsistency between the previous and current biopsies, the patient's former biopsy material was reevaluated. Histopathologically, most of the tumor consists of spindle schwannian component and mature ganglion cells. In one section of the first specimen, a cellular area of 1.5 mm diameter has been observed. The cells in this section are round blastic in appearance. In between these cells scattered mature ganglion cells were also seen. To exclude a lymphocytic infiltration, immunohistochemical staining was performed. These small cells were positive for CD56 and negative for LCA. The diagnosis of the first tumor was changed and accepted as a ganglioneuroblastoma (Fig. 2). The patient was discharged from the hospital with no neurological deficits. An anterior approach was planned for the resection of the retroperitoneal rest tumor and oncological treatment afterwards. However, the patient refused to have further treatment and lost to follow-up.
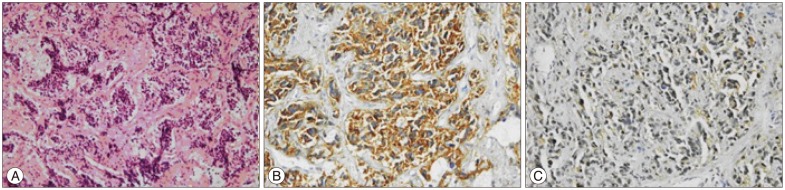
Nine years after the second operation, the patient admitted with progressive paraparesis, urinary retention and hypoesthesia. MRI revealed a right foraminal T10 extradural mass (Fig. 3). With posterior approach and T10 right hemilaminectomy the extradural part of soft, white-brown coloured tumor tissue within the spinal canal was removed. Neuropathological analysis showed a round cell malignant tumor without mature component. The tumor cells showed positive immunoreactivity for CD56, synaptophysin and negative immunoreactivity for desmin (Fig. 4). The histopathological diagnosis was NB. A genetic analysis was obtained. Interphase fluorescent in situ hybridisation (FISH) analysis was used for the evaluation of n-myc (2p24) status in the tumor cells with the use of "Cytocell n-myc amplification FISH probe". 1p and 11q deletions were assessed again with the interphase FISH analysis. "Vysis 1p36/1q25 and 19q13/19p13 FISH probe" and "Vysis 11q23 FISH probe" was used and it was shown that the n-myc oncogene was not amplified and there wer no deletions of chromosome 1p and 11q for all of the pathological samples.
DISCUSSION
Ganglioneuroma is a slow growing, benign tumor, which can be rarely associated with neurofibromatosis type I and multiple tumor syndromes like multiple endocrine neoplasia type IIB33). These tumors are fully differentiated tumors with mature ganglion cells and schwann cells16,23). Spinal ganglioneuromas represent less than 10% of all ganglioneuromas, usually with the involvement of paraspinal region28). Ganglioneuromas can remain asymptomatic for a long period and can be diagnosed incidentally. Surgical excision is the treatment of choice in the management of symptomatic ganglioneuromas with very good prognosis36). Ganglioneuroblastoma is one of the neuroblastic tumors and like ganglioneuroma or NB, it is also derived from the primordial neural crest cells. It contains both spindle schwannian stroma with mature ganglion cells and undifferentiated neuroblastic cells38).
NB is a tumor that primarily effects children. 89% of patients diagnosed with NB are under the age of 10 years13). Spinal involvement of NB constitues 20-30% of all pediatric primary extradural spinal tumors34). Familial genetic predisposititon and enviromental causes are thought to be responsible for the etiology of NB, although mechanisms leading to the disease are not fully understood5,12). NB is an unexpected pathological diagnosis in adults and spinal involvement of this tumor is extremely rare. There are limited number of cases with spinal NB in adults2,7,15,18,37). NB has a relatively poor prognosis in adults1,2).
The differentiation of NB into ganglioneuroblastoma or ganglioneuroma was first described by Cushing and Wolbach10) in 1927. Also the transformation from ganglioneuroma into malignant nerve sheath tumors was previously described3,8,14,17,20,31). However, the malignant transformation of a ganglioneuroma or a ganglioneuroblastoma into NB was only described by Kulkarni et al.22) in 1998. In this particular report malignant transformation from a ganglioneuroma to a NB was occured 11 years after the initial surgery. To our knowledge, it is the only report in the literature indicating such a condition.
The mechanism of the malignant transformation into NB is debatable. Kulkarni et al.22) proposed whether a malignant transformation of the remnant tumor or a neuroblastomatous component, which remained undiagnosed after the first operation. Considering the long survival period in both patients by Kulkarni et al. and the present report, namely 11 and 13 years, it is not possible to claim the latter proposal is realistic. However, with careful reevaluation of the former biopsy material, an undiagnosed cellular area with ganglioneuroblastomatous characteristics was found in our case. Four years after the first operation, it was observed that the whole tumor shared the pathological characteristics of this small ganglioblastomatous nodule. Although the biopsy of the first tumor in our case was in consistency with a ganglioneuroma, except this small nodule, it should be accepted as a ganglioneuroblastoma. After the second operation our patient was lost to follow-up, neither underwent surgery for the resection of the retroperitoneal rest tumor nor get any further oncological treatment. The total nine year survey after the second operation (a total of 13-year disease free survival until the neuroblastic transformation) despite a rest tumor and without any additional therapy is considerably long. Our case demonstrates that the biological behaviour of these tumors varies greatly and it might be more likely related to genetic factors rather than the histopathological characteristics.
Genetic inheritence of NB is also not fully understood. A polygenic inheritence is most probable. The limited number of familial cases limits the pursuit of genetic factors or familial inheritence in NB24). Well known genetic abnormalities in NB are chromosome deletion of 1p, 11q, gain of 17q and amplification of the n-myc oncogene6,27,32). Prior to the current report, there were no cytogenetic work-up has been done for the spinal NB. The specimens were tested for the amplification of the n-myc oncogene for the deletions of chromosome 1p and 11q. All these neurogenetic investigations were negative.
NB is known with its wide spectrum of clinical behavior. The prognosis might change in each particular case with regression, maturation, or progression of the tumors5,26). The variety in clinical behavior was found to be associated with spesific cytogenetic malformations9). Histologic grade, age, tumor differantiation, the status of n-myc oncogene, 11q and DNA ploidy are associated with clinical outcome9). Previously reportes cases of adult spinal NB is related with a shorter survival, less than 2 years2,18). However the present case and the case presented by Kulkarni et al.22) had similar survival of more than 10 years. This prolonged survival with stable disease may be related to better prognosis of spinal NB. For pediatric age group it is known that, spinal-paraspinal NB has a more favorable outcome with less frequent metastases21,29,35). Two genetic abnormalities, which were known to be oncogenic factors in NB, namely n-myc amplification and 1p, 11q deletions, were not found in this case. The relation between prolonged survival of our case and lack of this genetic abnormalities is debatable, however our patient is still doing well with no neurological deficits or complaints two years after the third operation with no recurrence.